Navigating RNA-seq Library Preparation Biases: A Comprehensive Guide for Robust Transcriptomic Analysis
RNA sequencing is a cornerstone of modern transcriptomics, yet its accuracy is fundamentally challenged by biases introduced during library preparation.
Navigating RNA-seq Library Preparation Biases: A Comprehensive Guide for Robust Transcriptomic Analysis
Abstract
RNA sequencing is a cornerstone of modern transcriptomics, yet its accuracy is fundamentally challenged by biases introduced during library preparation. This article provides a systematic guide for researchers and drug development professionals, exploring the foundational sources of bias from ligation to amplification, comparing methodological solutions for diverse applications including low-input and strand-specific sequencing, and offering troubleshooting strategies for optimization. By synthesizing evidence from recent comparative studies and technical evaluations, we outline a framework for validating library preparation methods to ensure data reliability, ultimately empowering robust experimental design and accurate biological interpretation in biomedical research.
Understanding the Roots: How Common Library Prep Steps Introduce Systematic Bias
Next-generation sequencing (NGS) has revolutionized biological research and clinical diagnostics. However, the intricate workflow of NGS, particularly for RNA sequencing (RNA-seq), introduces biases at nearly every step that can compromise data quality and lead to erroneous interpretation [1] [2]. A detailed understanding of these biases is essential for accurate data analysis and the development of improved protocols and bioinformatics tools. This technical support center provides a comprehensive guide to identifying, troubleshooting, and mitigating these pervasive biases in your NGS experiments.
Frequently Asked Questions (FAQs)
1. What are the most common sources of bias in RNA-seq library preparation? Biases can originate from nearly every step of the process. The most common sources include [2] [3]:
- Sample Preservation: RNA degradation during storage, especially in Formalin-Fixed Paraffin-Embedded (FFPE) samples, and cross-linking of nucleic acids with proteins.
- RNA Extraction: Inefficient purification or loss of specific RNA types (e.g., small RNAs) using certain methods like TRIzol.
- mRNA Enrichment: 3'-end capture bias during poly(A) selection and inefficiency in ribosomal RNA (rRNA) depletion.
- Fragmentation: Non-random fragmentation leading to reduced sequence complexity.
- Primer Bias: Inefficient priming or mispriming, particularly with random hexamers.
- Reverse Transcription: Inefficient or biased cDNA synthesis.
- Adapter Ligation: Substrate preferences of ligase enzymes causing sequence-dependent ligation efficiency.
- PCR Amplification: Preferential amplification of fragments with specific properties (e.g., neutral GC content), leading to uneven coverage and loss of library complexity.
2. How can I tell if my NGS library has a high degree of bias? Several indicators can signal a biased library [4]:
- Uneven coverage across the genome or transcriptome.
- High duplication rates in the sequencing data.
- Abnormal sequence content, such as significant AT or GC enrichment.
- The presence of adapter-dimers or other artifact sequences in quality control reports (e.g., a sharp peak at ~70-90 bp on a Bioanalyzer trace).
- Low library complexity, meaning a high number of PCR duplicates from a low number of original RNA molecules.
3. My sequencing run yielded very low coverage. What steps should I investigate first? Low yield can stem from multiple issues. A systematic troubleshooting approach is recommended [4]:
- Input Sample Quality: Verify RNA Integrity Number (RIN) or DNA quality. Check for contaminants (e.g., salts, phenol) that inhibit enzymes by ensuring good 260/230 and 260/280 ratios.
- Quantification: Use fluorometric methods (Qubit) instead of absorbance (NanoDrop) for accurate quantification of usable material.
- Fragmentation: Confirm that fragmentation produced the expected size distribution.
- Adapter Ligation: Ensure correct adapter-to-insert molar ratios and optimal ligation conditions.
- Purification: Review cleanup steps (e.g., bead ratios) to avoid accidental removal of the target library.
4. Are there PCR-free methods to avoid amplification bias? Yes, PCR-free protocols are available and are recommended when a large amount of high-quality input DNA is available [2]. These protocols circumvent PCR amplification by directly ligating adapters to the DNA fragments, thereby eliminating biases associated with unequal amplification. However, these methods require microgram quantities of input DNA and may still present other artifacts [2].
Troubleshooting Guides
Guide 1: Addressing PCR Amplification Bias
Problem: PCR amplification stochastically introduces biases, preferentially amplifying certain fragments over others. This leads to uneven coverage, loss of library complexity, and an overrepresentation of duplicates in sequencing data [2].
Solutions:
- Minimize PCR Cycles: Use the minimum number of PCR cycles necessary to generate sufficient library material [2].
- Polymerase Selection: Use high-fidelity polymerases, such as Kapa HiFi, which are designed for more uniform amplification compared to others like Phusion [2].
- PCR Additives: For extremely AT-rich or GC-rich sequences, use additives like TMAC (tetramethylammonium chloride) or betaine to help neutralize base-composition bias [2].
- PCR-Free Protocols: Whenever input material allows, adopt PCR-free library preparation methods [2].
Table 1: Troubleshooting PCR Amplification Bias
| Symptom | Possible Cause | Corrective Action |
|---|---|---|
| High duplicate read rate | Too many PCR cycles; low input complexity | Reduce PCR cycles; increase input material |
| Skewed coverage in GC-rich/AT-rich regions | Polymerase bias against extreme GC/AT content | Use PCR additives (TMAC, betaine); optimize extension temperature/time |
| Low library diversity | Preferential amplification of a subset of fragments | Switch polymerase (e.g., to Kapa HiFi); use unique molecular identifiers (UMIs) |
Guide 2: Mitigating Bias from RNA Input and Fragmentation
Problem: The quality, quantity, and fragmentation of the input RNA can significantly impact the representativeness of the final sequencing library. Degraded or low-input RNA reduces complexity, while non-random fragmentation creates length biases [2].
Solutions:
- Input RNA Quality: For degraded samples (e.g., FFPE), use high RNA input and switch to random priming for reverse transcription instead of oligo-dT [2].
- RNA Extraction Method: Select extraction methods optimized for your RNA species of interest. For example, the mirVana miRNA kit is reported to be superior for small RNA yield and quality compared to TRIzol [2].
- Fragmentation Method: Use chemical treatment (e.g., zinc-based fragmentation) rather than enzymatic methods (e.g., RNase III) for more random fragmentation [2]. Alternatively, fragment the cDNA after reverse transcription using mechanical or enzymatic methods [2].
Table 2: Troubleshooting RNA Input and Fragmentation Issues
| Symptom | Possible Cause | Corrective Action |
|---|---|---|
| Low mapping rates; 3'-bias in coverage | RNA degradation; use of oligo-dT on degraded RNA | Check RNA integrity (RIN); use random primers for RT |
| Loss of small RNA representation | Suboptimal RNA extraction method | Use specialized kits (e.g., mirVana) for small RNA isolation |
| Reduced sequence complexity | Non-random RNA fragmentation | Switch from enzymatic to chemical fragmentation methods |
Guide 3: Resolving Adapter Ligation and Primer Bias
Problem: The enzymes used in adapter ligation and reverse transcription can have sequence-dependent preferences, leading to the under-representation of certain sequences in your library [2].
Solutions:
- Adapter Design: Use adapters with random nucleotides at the ligation junctions. This helps to minimize the substrate preference of T4 RNA ligases [2].
- Ligation Conditions: Optimize adapter-to-insert molar ratios, temperature, and duration. For cohesive-end ligation, lower temperatures (12-16°C) and longer durations (overnight) can enhance efficiency, especially for low-input samples [5].
- Priming Strategy: To mitigate random hexamer priming bias, one proposed solution is to avoid converting RNA to double-stranded cDNA with random primers altogether. Instead, sequencing adapters can be ligated directly onto the RNA fragments themselves [2]. Bioinformatics tools can also be used to reweight read counts to adjust for this bias [2].
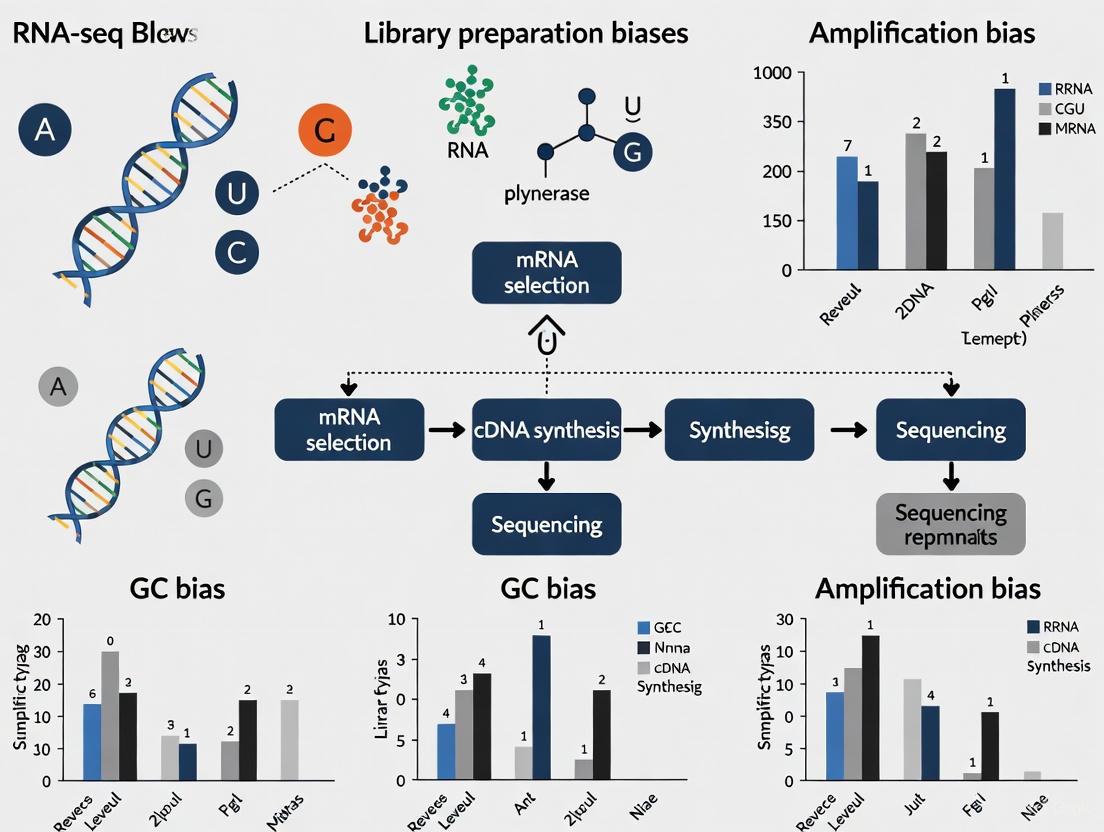
The following diagram illustrates a generalized NGS workflow for RNA-seq with key sources of bias highlighted at each stage.
Research Reagent Solutions
The following table lists key reagents and materials used in NGS library preparation, along with their functions and considerations for mitigating bias.
Table 3: Essential Reagents and Their Roles in Mitigating NGS Bias
| Reagent/Material | Function in Workflow | Considerations for Bias Reduction |
|---|---|---|
| RNA Extraction Kits (e.g., mirVana) | Isolate and purify RNA from samples. | Select kits validated for your RNA species of interest (e.g., small RNAs) to avoid selective loss [2]. |
| Oligo-dT Beads / rRNA Depletion Kits | Enrich for polyadenylated mRNA or remove ribosomal RNA. | Be aware of 3'-end capture bias with oligo-dT. Use rRNA depletion for non-polyadenylated transcripts or degraded RNA [2]. |
| Fragmentation Reagents (Enzymatic vs. Chemical) | Break RNA into appropriately sized fragments for sequencing. | Chemical fragmentation (e.g., zinc) is often more random than enzymatic (RNase III), reducing sequence-based bias [2]. |
| Reverse Transcriptase | Synthesize first-strand cDNA from RNA template. | Use high-efficiency enzymes. Consider random hexamer bias and explore alternative strategies like direct RNA ligation [2]. |
| Adapter Oligos | Provide sequences necessary for binding to the flow cell and indexing. | Use adapters with random base extensions at ligation ends to combat ligase sequence preference [2]. Ensure correct molar ratios to prevent adapter-dimer formation [5]. |
| High-Fidelity DNA Polymerase (e.g., Kapa HiFi) | Amplify the adapter-ligated library to generate sufficient mass for sequencing. | Selected for uniform amplification across sequences with varying GC content to minimize PCR bias [2]. |
| Solid Phase Reversible Immobilization (SPRI) Beads | Purify and size-select nucleic acid fragments between enzymatic steps. | Precisely control bead-to-sample ratios to avoid skewed size selection and loss of desired fragments [4]. |
In RNA sequencing (RNA-seq) library preparation, the ligation of adapter sequences to RNA fragments is a fundamental step that enables subsequent amplification and sequencing. However, this process is not neutral; T4 RNA ligases used in these procedures demonstrate strong sequence-specific biases and structure-specific preferences that systematically distort the representation of RNA species in the final sequencing library [6] [7]. This ligation bias originates from the inherent substrate preferences of the enzymes themselves, particularly T4 RNA Ligase 1 (Rnl1) and truncated T4 RNA Ligase 2 (Rnl2tr) [6] [8]. When certain RNA fragments ligate more efficiently than others due to their terminal sequences or structural features, the resulting sequencing data no longer accurately reflects the original RNA abundances, potentially leading to erroneous biological conclusions [2] [1]. Understanding and mitigating this bias is therefore crucial for any researcher relying on RNA-seq data for transcriptome analysis, small RNA discovery, or quantitative gene expression studies.
Mechanisms: How Ligation Bias Occurs
Ligation bias in RNA-seq libraries arises through two primary, interconnected mechanisms: sequence-specific preferences and structural constraints.
Sequence-Specific Bias of RNA Ligases
The RNA ligases used in library preparation do not treat all sequence ends equally. Comprehensive studies using randomized RNA pools have demonstrated that these enzymes have intrinsic preferences for certain nucleotides at positions near the ligation junction [6] [8]. One study found that the thermostable ligase Mth K97A exhibited a strong preference for adenine and cytosine at the third nucleotide from the ligation site [8]. This means that RNA fragments with preferred nucleotides at their ends will be overrepresented in the final library, while those with disfavored sequences will be underrepresented, creating a distorted view of the actual RNA population.
Structural Bias: RNA and Adaptor Co-folding
Beyond primary sequence, the secondary structure of RNA fragments and their ability to co-fold with adapter sequences significantly impacts ligation efficiency [7]. Research has shown that over-represented sequences in sequencing libraries are more likely to be predicted to have secondary structure and to co-fold with adaptor sequences [8] [7]. These structures can either facilitate or hinder the ligation reaction by making the RNA ends more or less accessible to the ligase enzyme. One investigation noted that "over-represented fragments were more likely to co-fold with the adaptor," suggesting that certain RNA-adaptor combinations form favorable structural contexts that promote efficient ligation [8].
The following diagram illustrates the key experimental findings on the sources and impacts of ligation bias:
Experimental Evidence and Quantitative Data
Multiple studies have systematically quantified the extent and impact of ligation bias in RNA-seq library preparation. The following table summarizes key experimental findings from the literature:
Table 1: Quantitative Evidence of Ligation Bias from Experimental Studies
| Study System | Experimental Approach | Key Finding on Bias Magnitude | Implication |
|---|---|---|---|
| Randomized RNA pool [8] | Comparison of library preparation protocols | Most abundant sequences were present ≥5 times more than expected without bias; CircLig protocol reduced over-representation by approximately half compared to standard protocol | Even the best available protocols significantly distort RNA representation |
| Defined miRNA mixtures [7] | Comparison of miRNA detection with different adaptors | Using randomized adaptors in both ligation steps produced HTS results that better reflected the starting miRNA pool | Adaptor sequence directly impacts quantification accuracy |
| Mouse insulinoma cells [9] | Comparison of Illumina v1.5 vs. TruSeq protocols | 102 highly expressed miRNAs were >5-fold differentially detected between protocols; some miRNAs (e.g., miR-24-3p) showed >30-fold differential detection | Choice of commercial library prep kit drastically affects results |
| Small RNA sequencing [6] | Testing of modified adaptor strategies | Identified reproducible discrepancies specifically arising from ligation or amplification steps, with T4 RNA ligases as the predominant cause of distortions | Points to a specific enzymatic step as the primary source of bias |
The evidence consistently demonstrates that ligation bias is not a minor technical artifact but a substantial factor that can dramatically alter the perceived abundance of RNA species, potentially leading to incorrect biological interpretations.
Troubleshooting Guide: Addressing Ligation Problems
Researchers encountering potential ligation bias in their RNA-seq experiments can use the following troubleshooting guide to identify and resolve common issues:
Table 2: Troubleshooting Guide for Ligation-Related Issues in RNA-Seq
| Problem | Potential Causes | Recommended Solutions | Supporting Evidence |
|---|---|---|---|
| Uneven RNA representation | Sequence-specific ligase preference; RNA secondary structure | Use pooled adaptors with random nucleotides at ligation boundaries; employ structured adaptors that promote uniform ligation | Demonstrated to recover miRNAs that evade capture by standard methods [9] and reduce bias [6] [7] |
| Low library diversity | Inefficient ligation of certain RNA species; high adapter dimer formation | Use chemically modified adapters that inhibit dimer formation; optimize adapter concentration and purification steps | Kits addressing adapter dimers produce higher-quality results with lower input RNA [9] |
| Inconsistent results between protocols | Different adapter sequences and ligation conditions | Standardize library preparation method across experiments; when comparing datasets, account for protocol differences bioinformatically | Different Illumina protocols produced strikingly different miRNA profiles from the same RNA sample [9] |
| Poor ligation efficiency | Enzyme inhibitors; suboptimal reaction conditions | Ensure RNA is free of contaminants (salts, EDTA, phenol); use fresh ATP-containing buffer; optimize enzyme concentration and incubation time | Reaction efficiency decreases with degraded ATP and inhibitors [10] [11] |
Frequently Asked Questions (FAQs)
Q1: Why do different commercial library preparation kits produce different results from the same RNA sample? Different kits use distinct adapter sequences and ligation conditions, which interact variably with the diverse sequences and structures in your RNA population. Studies have shown that these differences can cause >30-fold variation in the detection of some miRNAs [9]. This occurs because each adapter sequence has different ligation efficiencies with different RNA ends, and each ligase enzyme has its own sequence and structure preferences [6] [7].
Q2: Can I bioinformatically correct for ligation bias after sequencing? While some bioinformatic methods exist to partially compensate for ligation bias, such as read count reweighing schemes [2], they cannot fully eliminate bias introduced during the physical library preparation process. The most effective approach combines wet-lab biochemical optimizations (like using pooled adapters) with bioinformatic corrections, as post-sequencing corrections cannot recover RNAs that completely failed to ligate during library preparation [8] [9].
Q3: How does RNA quality affect ligation bias? RNA quality significantly impacts ligation efficiency and bias. Degraded RNA with fragmented ends presents diverse terminal sequences that may ligate with varying efficiencies, increasing bias [2] [12]. High-quality RNA with minimal degradation provides more consistent ligation substrates. Always quality-check RNA using methods like Bioanalyzer/TapeStation (RIN >8 recommended) and use nuclease-free techniques to prevent degradation [12] [9].
Q4: Are there specific RNA types more susceptible to ligation bias? Yes, small RNAs with significant secondary structure near their termini are particularly prone to ligation bias because structure affects adapter accessibility [7]. Some miRNAs with specific terminal sequences may be consistently under-represented with certain adapter sets [9]. RNAs with extreme GC content may also exhibit biased representation due to structural constraints and melting temperature considerations during ligation [2] [8].
Research Reagent Solutions
The following table catalogues key reagents and methodologies discussed in the literature for mitigating ligation bias:
Table 3: Research Reagents and Methods for Reducing Ligation Bias
| Reagent/Method | Purpose/Function | Evidence of Efficacy |
|---|---|---|
| Pooled Adapters (e.g., NEXTflex V2) | Adapters with random nucleotides at ligation boundaries provide diverse ligation contexts | Detects miRNAs missed by standard methods; correlates better with RT-qPCR data [9] |
| trRnl2 K227Q mutant | Reduced bias variant of T4 RNA Ligase 2 | Associated with almost half the level of over-representation compared to standard protocol [8] |
| CircLigase-based protocol | Single adaptor approach that avoids T4 Rnl1 | Results in less over-representation of specific sequences than standard protocol [8] |
| Structured Adapters | Adapters with complementary regions that promote uniform circularization | Encourages consistent structural context for all miRNAs, reducing bias [7] [9] |
| Chemical modification of adapters | Prevents adapter dimer formation | Increases proportion of informative sequencing reads, especially critical for low-input samples [9] |
Experimental Protocol: Pooled Adapter Strategy for Bias Reduction
This protocol is adapted from studies that successfully reduced ligation bias using adapter pooling strategies [6] [9]. The following workflow diagram illustrates the key steps:
Adapter Design and Synthesis
- Design 5' and 3' adapters with 2-4 random nucleotides (NN or NNNN) at the ligation boundaries [6] [9].
- For additional bias reduction, consider designing adapters with short complementary regions that encourage formation of uniform structures during ligation [7].
- Synthesize the adapter pools using mixed base chemistry to ensure diversity.
- For 3' DNA adapters, include 5' rAPP and 3'ddC modifications to prevent self-ligation and circularization [6].
Library Preparation Steps
- Starting Material: Use high-quality, high-integrity RNA (RIN >8) to minimize additional sources of bias [12] [9].
- 3' Adapter Ligation:
- Ligate the pooled 3' adapter to RNA using truncated T4 RNA Ligase 2 (Rnl2tr) or the bias-reduced mutant K227Q in an appropriate buffer.
- Use ATP-free conditions for this step when using pre-adenylated adapters [6].
- Purification: Purify the ligation products by denaturing PAGE to precisely isolate RNA-adapter conjugates and remove excess adapter [6] [9].
- 5' Adapter Ligation:
- Ligate the pooled 5' adapter using T4 RNA Ligase 1 (Rnl1) with ATP-containing buffer [6].
- Reverse Transcription and Amplification:
- Final Purification: Gel-purify the completed library to remove PCR artifacts and adapter dimers.
Data Analysis Considerations
- Pre-processing: Trim the random bases from the beginning and end of sequencing reads before alignment [9].
- Quality Assessment: Compare the distribution of detected RNAs to expected abundances when working with defined samples.
- Validation: Consider validating key findings with an orthogonal method such as RT-qPCR, especially for RNAs of particular biological interest [9].
This protocol leverages the principle that providing diverse adapter sequences increases the probability that each RNA species will encounter an adapter with which it can ligate efficiently, thereby producing a more representative library that better reflects the true composition of the original RNA sample [6] [7] [9].
Ribosomal RNA (rRNA) constitutes a formidable challenge in transcriptome studies, representing over 80-90% of total RNA in most cells [13] [14] [15]. This overwhelming abundance necessitates efficient removal or enrichment strategies to enable meaningful sequencing of informative RNA species. The two predominant methods for addressing this challenge—polyA+ selection and rRNA depletion (ribodepletion)—employ fundamentally different principles, each introducing specific biases and technical considerations that impact downstream data interpretation [2] [14].
Within the broader context of research on RNA-seq library preparation biases, understanding the methodological choice between these approaches becomes paramount. This technical support center document synthesizes current evidence to guide researchers in selecting appropriate protocols, troubleshooting common issues, and implementing best practices tailored to their experimental goals, sample types, and biological questions.
Core Technology Comparison: Mechanisms and Technical Specifications
Fundamental Mechanisms of Action
PolyA+ Selection utilizes oligo-dT primers or beads to hybridize to the polyadenylated 3' tails of mature messenger RNAs (mRNAs) [16] [17]. This mechanism selectively enriches for polyadenylated transcripts while excluding rRNAs, transfer RNAs (tRNAs), and other non-polyadenylated species. This method provides a targeted approach but is inherently limited to transcripts containing intact polyA tails.
rRNA Depletion (Ribodepletion) employs sequence-specific DNA or RNA probes complementary to ribosomal RNA sequences [18] [16]. These probes hybridize to rRNA molecules, which are subsequently removed from the total RNA pool through magnetic bead capture or enzymatic digestion. This strategy preserves both polyadenylated and non-polyadenylated transcripts, offering a broader view of the transcriptome.
The workflow for each method can be visualized as follows:
Quantitative Performance Comparison
The choice between polyA+ selection and rRNA depletion significantly impacts key sequencing metrics and data quality. Performance varies substantially across sample types, RNA integrity levels, and target organisms.
Table 1: Comparative Performance of rRNA Depletion vs. PolyA+ Selection Across Sample Types
| Performance Metric | Blood Samples | Colon Tissue | FFPE Samples | Bacterial Samples |
|---|---|---|---|---|
| Usable Exonic Reads | 22% (rRNA depletion) vs. 71% (polyA+) [14] | 46% (rRNA depletion) vs. 70% (polyA+) [14] | ~20% (rRNA depletion) [15] | Highly variable by depletion method [18] |
| Intronic/Intergenic Reads | ~62% (rRNA depletion) vs. ~32% (polyA+) [15] | Similar pattern as blood but less pronounced [14] | >60% (rRNA depletion) [15] | Not applicable |
| Additional Reads Needed for Equivalent Exonic Coverage | 220% more with rRNA depletion [14] | 50% more with rRNA depletion [14] | Protocol dependent [15] | Method dependent [18] |
| rRNA Removal Efficiency | Up to 97-99% with optimized probes [16] | Up to 97-99% with optimized probes [16] | Comparable to polyA+ in fresh-frozen [15] | Varies by kit: 65-95% [18] |
Table 2: Transcript Detection Capabilities by RNA Biotype
| RNA Biotype | PolyA+ Selection | rRNA Depletion | Key Implications |
|---|---|---|---|
| Protein-coding mRNA | High detection efficiency [14] | High detection efficiency [14] | Both methods suitable for coding transcripts |
| Long non-coding RNA (lncRNA) | Limited to polyadenylated forms [14] | Comprehensive detection [13] [14] | rRNA depletion essential for complete lncRNA profiling |
| Histone mRNAs | Not detected (lack polyA tails) [17] | Detected [17] | Critical consideration for epigenetics studies |
| Pre-mRNA & Nascent Transcripts | Minimal detection [14] | Significant detection [14] [15] | rRNA depletion enables analysis of transcriptional regulation |
| Small RNAs | Not efficiently captured [14] | Detected but may require specific protocols [14] | Specialized small RNA protocols recommended |
| Non-polyadenylated Viral RNAs | Not detected [17] | Detected [17] | Important for virology and pathogen discovery |
Troubleshooting Guides: Addressing Common Experimental Challenges
Library Preparation and Quality Control Issues
Observation: High adapter-dimer peaks (~127 bp) on Bioanalyzer
- Possible Causes: Addition of undiluted adapter; RNA input too low; RNA over-fragmented or lost during fragmentation; inefficient ligation [19].
- Effects: Adapter-dimer will cluster and be sequenced, wasting sequencing capacity.
- Solutions: Dilute adaptor (10-fold dilution) before setting up ligation reaction; clean up PCR reaction again with 0.9X SPRIselect Beads (note: second clean up may reduce library yield) [19].
Observation: Additional Bioanalyzer peak at higher molecular weight (~1,000 bp)
- Possible Causes: PCR artifact from over-amplification [19].
- Effects: If ratio is low compared to library, may not problematic for sequencing.
- Solutions: Reduce number of PCR cycles to prevent primers from becoming limiting in late cycles [19].
Observation: Broad library size distribution
- Possible Causes: Under-fragmentation of RNA [19].
- Effects: Library will contain longer insert sizes, potentially affecting sequencing efficiency.
- Solutions: Increase RNA fragmentation time [19].
Method-Specific Performance Issues
Problem: High residual rRNA in ribodepletion libraries
- Potential Cause: Probe mismatch, especially in non-model organisms or with pan-prokaryotic kits [13] [18] [17].
- Solutions: Use species-specific probes; for non-model organisms, consider custom-designed probes [13] [18]; pilot a few samples and check percent rRNA before scaling [17].
- Technical Note: One study developed a custom set of 200 probes specifically matching C. elegans rRNA sequences, which significantly improved depletion efficiency compared to mammalian-optimized probes [13].
Problem: Strong 3' bias in polyA+ selected libraries
- Potential Cause: RNA degradation, particularly common in FFPE or clinically derived samples [15] [17].
- Solutions: For degraded samples, switch to rRNA depletion rather than increasing sequencing depth; use RNA integrity metrics (RIN/DV200) to guide method selection [17].
- Technical Note: rRNA depletion demonstrates more uniform coverage along transcript bodies and preserves 5' coverage better in compromised RNA [15] [17].
Problem: Low detection of non-coding RNAs
- Potential Cause: Using polyA+ selection, which excludes non-polyadenylated transcripts [13] [14].
- Solutions: Implement rRNA depletion to capture non-polyadenylated species including many lncRNAs, pre-processed RNAs, and regulatory RNAs [13] [16].
Frequently Asked Questions (FAQs)
Q1: When should I choose polyA+ selection versus rRNA depletion for my RNA-seq experiment?
A: The decision should be guided by three key factors: (1) organism, (2) RNA integrity, and (3) research focus [17]:
- Choose polyA+ selection for: Intact eukaryotic RNA (RIN ≥7 or DV200 ≥50%); primary focus on coding mRNA; gene-level differential expression studies [14] [17].
- Choose rRNA depletion for: Degraded or FFPE samples; need to detect non-polyadenylated RNAs (lncRNAs, histone mRNAs, viral RNAs); prokaryotic transcriptomics; studies requiring information on nascent transcription [15] [16] [17].
Q2: How does RNA quality impact method selection?
A: RNA quality significantly affects performance:
- High-quality RNA: Both methods work well, with polyA+ selection providing higher exonic mapping rates for coding genes [14].
- Degraded/FFPE RNA: rRNA depletion outperforms polyA+ selection due to better tolerance of fragmentation and crosslinks [15] [17]. PolyA+ selection on degraded RNA produces strong 3' bias and under-represents long transcripts [17].
Q3: What are the key differences in data output between these methods?
A: The methods produce fundamentally different data profiles:
- PolyA+ selection: Higher percentage of reads mapping to exonic regions; more efficient for gene-level quantification of protein-coding genes; lower sequencing depth required for equivalent exon coverage [14].
- rRNA depletion: Captures wider diversity of transcript types; higher percentage of intronic reads (indicative of pre-mRNA); requires more sequencing depth to achieve equivalent exon coverage; enables detection of non-polyadenylated RNAs [13] [14] [15].
Q4: Are there organism-specific considerations for rRNA depletion?
A: Yes, organism-specific optimization is critical:
- Prokaryotes: PolyA+ selection is not appropriate; rRNA depletion or targeted capture is standard [18] [17].
- Non-model eukaryotes: Commercial pan-eukaryotic kits may have suboptimal efficiency; custom probes designed against specific rRNA sequences significantly improve performance [13].
- Clinical samples: Blood samples may require additional globin RNA depletion alongside rRNA removal for optimal detection of low-expression transcripts [20].
Q5: How can I improve my ribodepletion efficiency?
A: Several strategies can enhance depletion:
- Use species-specific probes rather than pan-species kits [13] [18].
- Validate probe matching through pilot studies before scaling [17].
- For bacteria, consider methods that target 5S rRNA in addition to 16S and 23S rRNA [18].
- Ensure high-quality total RNA input through proper extraction and handling [16].
Essential Protocols and Methodologies
Experimental Workflow for Method Comparison
For researchers conducting comparative evaluations of polyA+ selection versus ribodepletion, the following experimental design provides a robust framework:
Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for rRNA Depletion and PolyA+ Selection Studies
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| rRNA Depletion Kits | Ribo-Zero Gold, RiboMinus, riboPOOLs, SoLo Ovation [13] [18] [15] | Remove ribosomal RNA via hybridization and magnetic bead capture; efficiency varies by species specificity [18]. |
| PolyA+ Selection Kits | SMARTSeq V4, TruSeq RNA Library Prep Kit [13] [21] | Enrich polyadenylated transcripts using oligo-dT primers or beads; optimized for eukaryotic mRNA [14]. |
| Custom Probe Design Services | Tecan Genomics Custom AnyDeplete, self-designed biotinylated probes [13] [18] | Species-specific rRNA depletion for non-model organisms; significantly improves efficiency [13]. |
| Library Preparation Systems | NEBNext Ultra Directional RNA Kit, TruSeq Stranded Total RNA Kit [19] [21] | Post-enrichment/depletion library construction; impact strand-specificity and bias patterns [2]. |
| RNA Quality Assessment Tools | Agilent Bioanalyzer, Qubit Fluorometer, RNA Integrity Number (RIN) [13] [21] | Critical for method selection; determines suitability for polyA+ selection [17]. |
| Bias Reduction Additives | Kapa HiFi Polymerase, PCR additives (TMAC, betaine) [2] | Reduce GC bias and improve amplification uniformity; particularly important for extreme GC content genomes [2]. |
The choice between polyA+ selection and rRNA depletion represents a fundamental experimental design decision that shapes all subsequent data interpretation. Within the broader research context of RNA-seq library preparation biases, evidence consistently demonstrates that each method offers distinct advantages and limitations that must be aligned with research objectives.
For coding transcript quantification in eukaryotes with high-quality RNA, polyA+ selection provides superior efficiency and exonic coverage. For comprehensive transcriptome characterization, including non-polyadenylated species, or when working with degraded clinical samples or prokaryotes, rRNA depletion offers the necessary breadth and tolerance. Critically, custom species-specific probe design significantly enhances ribodepletion performance, particularly for non-model organisms [13] [18].
As transcriptomics continues to evolve toward more nuanced applications—including single-cell sequencing, spatial transcriptomics, and multi-omics integration—understanding these foundational methodological choices remains essential for generating biologically meaningful, technically robust data that advances both basic research and drug development.
PCR amplification is a fundamental step in preparing DNA and RNA sequencing libraries. However, this process introduces various artifacts that can significantly skew quantitative measurements in your data. Understanding these artifacts—including PCR duplicates, amplification bias, and mispriming events—is crucial for accurate interpretation of sequencing results, particularly in quantitative applications like gene expression analysis [22] [23] [24].
This guide addresses the most common PCR-related artifacts, their impact on data integrity, and provides practical solutions for identification and troubleshooting within the context of RNA-seq library preparation bias research.
Frequently Asked Questions
What are PCR duplicates and how do they affect my RNA-seq data?
PCR duplicates are multiple identical reads originating from a single original DNA or RNA fragment due to PCR amplification [23]. In RNA-seq experiments, they can artificially inflate counts for specific transcripts, leading to inaccurate gene expression measurements [25] [26]. However, it's important to distinguish true PCR duplicates from "natural duplicates" (multiple independent fragments from highly expressed genes), as removing the latter can introduce bias [26].
How can I identify if PCR artifacts are affecting my data?
Several indicators suggest PCR artifact contamination:
- Inability to verify RNA-seq results with qPCR (failure to confirm 18 out of 20 differentially expressed genes) [25]
- High percentages of identical reads (approximately 25% reads removed during deduplication) [25]
- Recurrent ambiguous base calls at specific genomic positions [22]
- Spurious peaks with flush 3' ends in short RNA-seq data [24]
What causes uneven amplification across different sequences?
Multi-template PCR often results in non-homogeneous amplification due to sequence-specific factors [27]. Even with a slight (5%) amplification efficiency disadvantage relative to other templates, a sequence can be underrepresented by approximately two-fold after just 12 PCR cycles [27]. This bias occurs due to:
- Sequence-specific amplification efficiencies independent of GC content
- Adapter-mediated self-priming mechanisms
- Primer binding site mutations in evolving templates like viruses [22]
Should I always remove duplicate reads from RNA-seq data?
Not necessarily. For RNA-seq data, 70-95% of read duplicates may be "natural duplicates" from highly expressed genes rather than technical PCR duplicates [26]. Removing these natural duplicates can bias expression quantification. Computational methods that leverage heterozygous variants can help distinguish between natural and PCR duplicates for accurate estimation of true PCR duplication rates [26].
Troubleshooting Guide
Problem: Suspected PCR Artifacts in RNA-seq Data
Symptoms:
- Discrepancy between RNA-seq and qPCR validation results [25]
- Unexplained differential expression with high duplicate reads [25]
- Spurious peaks or inconsistent coverage across samples [24]
Solutions:
- Computational Assessment: Use tools like
PCRduplicatesto estimate true PCR duplication rates by leveraging heterozygous variants in your data [26]. - Sequence Analysis: Check for flush 3' ends in read alignments and complementarity to RT-primer sequences, which may indicate reverse transcription mispriming [24].
- Deduplication Strategy: Apply appropriate deduplication based on your experiment type. For DNA-seq, most duplicates are technical, while for RNA-seq, most may be biological [26].
Problem: Primer-Related Artifacts in Targeted Sequencing
Symptoms:
- Recurrent ambiguous base calls or consistent base calling errors [22]
- Amplicon drop-outs in specific regions [22]
- Rapid accumulation of ambiguous bases during new variant emergence [22]
Solutions:
- Primer Scheme Updates: Continuously monitor and update primer schemes to address viral evolution [22].
- Spike-in Primers: Implement custom "spike-in" primers for emerging variants with primer binding site mutations [22].
- Reference Genome Optimization: Use reference genomes with closer genetic distance to your samples to improve read mapping [22].
Problem: Non-Homogeneous Amplification in Multi-template PCR
Symptoms:
- Progressive skewing of coverage distributions with increased PCR cycles [27]
- Severe depletion of specific amplicon sequences [27]
- Inaccurate abundance measurements in quantitative applications [27]
Solutions:
- Cycle Optimization: Minimize PCR cycles to reduce amplification skew while maintaining sufficient library complexity [23].
- Deep Learning Prediction: Utilize computational models (1D-CNNs) to predict sequence-specific amplification efficiencies during experimental design [27].
- Alternative Enzymes: Consider thermostable group II intron-derived reverse transcriptases (TGIRT) to avoid mispriming artifacts [24].
Quantitative Impact of PCR Artifacts
Table 1: Common PCR Artifacts and Their Quantitative Impacts
| Artifact Type | Primary Cause | Effect on Quantitative Data | Typical Frequency |
|---|---|---|---|
| PCR Duplicates | Over-amplification of original fragments | Artificial inflation of read counts for specific sequences | 5-30% in RNA-seq [26] |
| Amplification Bias | Sequence-specific efficiency differences | Skewed abundance measurements; under-representation of low-efficiency templates | 2% of sequences with very poor efficiency (<80% of mean) [27] |
| Primer-Index Hopping | Mismatches in primer binding sites | Ambiguous base calls; amplicon drop-outs; omitted defining mutations | Rapid accumulation during variant emergence [22] |
| RT Mispriming | Non-specific annealing of RT primers | False cDNA ends; spurious peaks in coding exons | 10,000+ sites per dataset in affected studies [24] |
Table 2: Comparison of Computational Detection Methods for PCR Artifacts
| Method | Target Artifact | Key Features | Limitations |
|---|---|---|---|
| Heterozygous Variant Analysis [26] | PCR duplicates | Distinguishes technical vs. natural duplicates; Works on existing data | Requires heterozygous sites in genome |
| Mispriming Identification Pipeline [24] | RT mispriming | Identifies artifacts with minimal complementarity (2 bases); Filters spurious peaks | Requires specific sequence patterns |
| Deduplication Tools | PCR duplicates | Standard in most pipelines; Reduces redundant reads | Risk of removing biological duplicates in RNA-seq |
| Deep Learning Models [27] | Amplification bias | Predicts efficiency from sequence alone; Designs homogeneous libraries | Requires training data; Complex implementation |
Experimental Protocols
Protocol 1: Estimating True PCR Duplication Rate from Sequence Data
This protocol estimates the PCR duplication rate while accounting for natural duplicates using heterozygous variants [26].
- Identify Duplicate Clusters: Group reads with identical outer mapping coordinates.
- Select Heterozygous Sites: Use known or called heterozygous SNVs in your sample.
- Analyze Allele Patterns: For duplicate clusters of size 2 overlapping heterozygous sites:
- Clusters with opposite alleles indicate natural duplicates
- Clusters with identical alleles indicate PCR duplicates
- Calculate Proportions: Estimate unique DNA fragments using the formula:
- U₂ = [1·(C₂ - 2C₂₁) + 2·2C₂₁] / C₂
- Where C₂ = total clusters of size 2, C₂₁ = clusters with opposite alleles
- Extend to Larger Clusters: Apply mathematical modeling to estimate proportions for larger cluster sizes.
Protocol 2: Identifying and Removing RT Mispriming Artifacts
This computational pipeline identifies cDNA reads produced from reverse transcription mispriming [24].
- Read Alignment: Use a global aligner (BWA) for sequencing reads.
- Filter Non-coding RNAs: Remove reads mapping to known non-protein-coding genes.
- Identify Flush-ended Peaks: Find genomic positions with >10 reads having identical 3' ends.
- Check Adapter Complementarity: Identify peaks adjacent to:
- Dinucleotides matching the 3' adapter (k-mer sites)
- Dinucleotides not matching the 3' adapter (non-k-mer sites)
- Apply Mispriming Criteria: Classify as mispriming sites if:
- At least two bases match the 3' end of the 3' adapter
- No non-k-mer site with similar flush ends within 20 bases
- Filter Artifactual Reads: Remove reads identified as mispriming artifacts.
Experimental Workflow: From RNA Extraction to Artifact Detection
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 3: Essential Reagents and Methods for Managing PCR Artifacts
| Reagent/Method | Primary Function | Advantages | Considerations |
|---|---|---|---|
| Unique Molecular Identifiers (UMIs) [26] | Tags individual molecules before amplification | Enables precise distinction of PCR duplicates from natural duplicates | Requires specialized library prep modifications |
| Low-Bias Library Prep Kits [28] | Reduces sequence-dependent amplification bias | Novel splint adaptor design; Broader input range; Simplified protocol | Commercial solution with associated costs |
| RiboGone Depletion Kits [29] | Removes ribosomal RNA from samples | Essential for random-primed protocols; Reduces wasteful rRNA sequencing | Specific to mammalian RNA in this variant |
| Thermostable Group II Intron RT (TGIRT) [24] | Reverse transcription with reduced mispriming | Template-switching activity avoids mispriming; Higher fidelity | Alternative enzyme system requiring protocol adjustment |
| SMARTer Stranded RNA-Seq Kit [29] | Maintains strand information with random priming | Works with degraded RNA (FFPE samples); >99% strand accuracy | Requires rRNA depletion or polyA selection |
| NEBNext Low-bias Small RNA Library Prep Kit [28] | Minimizes bias in small RNA representation | Fast protocol (3.5 hours); Broad input range; Handles 2'-O-methylated RNA | Specifically optimized for small RNA species |
| Artic Network Primers with Spike-ins [22] | Targeted enrichment for viral sequencing | Customizable for emerging variants; Continuously updated designs | Specific to viral genome applications |
Relationship Between PCR Cycles, Efficiency, and Coverage Skew
Effective management of PCR amplification artifacts requires both experimental and computational approaches. By implementing the detection methods and mitigation strategies outlined in this guide, researchers can significantly improve the quantitative accuracy of their sequencing data and draw more reliable biological conclusions.
Troubleshooting Guides
FAQ: Addressing Common Experimental Issues
Q1: My RNA-seq data shows uneven coverage, with poor representation of transcript ends. What could be the cause and how can I fix it?
Uneven coverage often stems from RNA fragmentation bias. Using RNase III for fragmentation is not completely random and can reduce sequence complexity [2]. To resolve this:
- Switch fragmentation methods: Use chemical treatment (e.g., zinc) instead of enzymatic digestion for a more random fragmentation pattern [2].
- Fragment after cDNA synthesis: Reverse transcribe intact RNA into cDNA first, then fragment the cDNA using mechanical or enzymatic methods [2].
- Verify with QC: Use quality control tools like RNA-SeQC to check for 3'/5' bias in your coverage metrics [30].
Q2: My library preparation seems to have a strong priming bias. How can I make my library more representative?
Priming bias, often from random hexamer mispriming, can skew transcript representation [2]. To mitigate this:
- Bypass random priming: For some protocols, you can ligate sequencing adapters directly onto RNA fragments, avoiding the cDNA synthesis step that uses random hexamers [2].
- Use a read count reweighing scheme: Apply computational correction post-sequencing to adjust for the bias and create a more uniform read distribution [2].
- Optimize primer choice and temperature: For gene-specific primers, ensure the sequence is unique to the target. Performing reverse transcription at a higher temperature with a thermostable enzyme can also increase primer binding specificity [31].
Q3: I am working with degraded RNA samples (e.g., FFPE). How can I minimize biases during library prep?
Degraded RNA requires specific adjustments to counteract biases introduced by fragmentation and priming [2]:
- Use random primers: Instead of oligo(dT) primers, which require an intact poly-A tail, use random hexamers for cDNA synthesis to ensure coverage across the entire fragment length of degraded RNA [31].
- Increase input material: Use a higher amount of starting RNA to compensate for degradation [2].
- Select a bias-resistant protocol: Choose a reverse transcriptase that works efficiently with degraded samples and consider PCR additives like betaine for AT/GC-rich regions [2] [31].
Table 1: Common Biases, Their Effects, and Quantitative Improvement Methods
| Bias Type | Effect on Coverage Uniformity | Improvement Method | Key Metric for QC |
|---|---|---|---|
| RNA Fragmentation Bias [2] | Reduced complexity; non-random fragment start/end sites. | Use chemical (e.g., zinc) instead of RNase III fragmentation [2]. | Coverage continuity; gap metrics (RNA-SeQC) [30]. |
| Random Hexamer Priming Bias [2] | Non-uniform read start sites; under-representation of transcript 5' ends. | Use direct RNA adapter ligation or read count reweighing [2]. | Uniformity of read start site distribution. |
| Adapter Ligation Bias [2] | Under-representation of sequences difficult to ligate. | Use adapters with random nucleotides at ligation extremities [2]. | Ligation efficiency measured by percentage of usable reads. |
| PCR Amplification Bias [2] | Preferential amplification of cDNA with neutral GC content; distortion of abundance. | Use polymerases like Kapa HiFi; reduce PCR cycles; additives (TMAC/betaine) for extreme GC% [2]. | Duplication rate; GC bias curve (RNA-SeQC) [30]. |
Experimental Protocols for Bias Mitigation
Protocol 1: Minimizing Fragmentation Bias with Chemical Treatment This protocol replaces enzymatic RNA fragmentation with divalent metal cations to generate more random fragments [2].
- Input: 1 µg of high-quality total RNA.
- Fragmentation Buffer Preparation: Prepare a 100 mM Zinc Chloride (ZnCl₂) solution in nuclease-free water.
- Reaction Setup: Combine RNA and fragmentation buffer to a final concentration of 10 mM ZnCl₂. Incubate at 70°C for 5-15 minutes (requires optimization for desired fragment size).
- Reaction Stop: Add 10 mM EDTA to chelate zinc and stop the reaction.
- Purification: Purify the fragmented RNA using RNA clean-up beads or columns. Proceed to library construction.
Protocol 2: Computational Correction for Random Hexamer Bias This bioinformatics protocol adjusts for non-uniform priming [2].
- Input Data: A BAM file containing aligned RNA-seq reads.
- Identify Primer Start Sites: Parse the alignment file to map the genomic positions corresponding to the start of each read (the presumed random hexamer binding site).
- Calculate Weighting Factors: For each transcript, model the expected uniform distribution of read starts. Calculate a weight for each read based on the observed vs. expected frequency of start sites in its region.
- Reweigh Read Counts: Apply the calculated weights to the read counts during transcript quantification.
- Output: A corrected count matrix for downstream differential expression analysis.
Workflow Diagrams
Diagram 2: Strategies for Bias Mitigation
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Bias Mitigation
| Reagent / Tool | Function | Role in Mitigating Bias |
|---|---|---|
| Zinc Chloride (ZnCl₂) [2] | Chemical RNA fragmentation agent. | Provides a more random fragmentation pattern compared to enzymatic methods like RNase III, reducing fragmentation bias and improving coverage uniformity. |
| Random Hexamers with Random Nucleotide Adapters [2] | Primers for cDNA synthesis and adapters for ligation. | Adapters with random nucleotides at ligation ends reduce substrate preference of ligases, mitigating adapter ligation bias. |
| Kapa HiFi Polymerase [2] | Enzyme for PCR amplification of libraries. | Reduces PCR amplification bias by offering superior performance and less GC-content preference compared to other polymerases like Phusion. |
| Betaine or TMAC [2] | PCR additives. | Helps neutralize extreme GC or AT content in templates, allowing for more uniform amplification of sequences with varying GC content. |
| RNA-SeQC [30] | Bioinformatics software for quality control. | Provides key metrics (like 3'/5' bias, coverage continuity, GC bias) to detect and quantify the presence of biases in the final dataset, informing inclusion criteria. |
| Thermostable Reverse Transcriptase [31] | Enzyme for synthesizing cDNA from RNA. | Withstands higher reaction temperatures, minimizing RNA secondary structures that cause reverse transcription bias and lead to truncated cDNA and poor coverage. |
Choosing Your Arsenal: Comparative Analysis of Library Prep Kits and Strategies
Key Comparisons at a Glance
The table below summarizes the core technical and practical differences between stranded and non-stranded RNA-seq protocols to guide your experimental design.
| Feature | Stranded RNA-seq | Non-Stranded RNA-seq |
|---|---|---|
| Protocol Complexity | More complex; additional steps (e.g., dUTP marking, strand degradation) [32] [33] | Simpler and more straightforward [32] [33] |
| Strand Information | Preserved. Determines if a read is from the sense or antisense DNA strand [32] | Lost. Cannot determine the transcript's strand of origin [32] |
| Quantitative Accuracy | More accurate, especially for genes with overlapping genomic loci on opposite strands [34] | Less accurate for overlapping genes; can misassign reads [34] |
| Read Ambiguity | Lower (~2.94% of reads ambiguous) [34] | Higher (~6.1% of reads ambiguous) [34] |
| Ideal Applications | Novel transcript/antisense discovery, genome annotation, complex transcriptome analysis [32] [33] | Gene expression profiling in well-annotated organisms, large-scale studies [32] |
| Cost & Input | Generally higher cost and may require more input RNA [35] | More cost-effective and can work with lower input amounts [35] [33] |
Experimental Protocols in Practice
Core Methodologies for Stranded RNA-seq
The dUTP second-strand marking method is a leading and widely adopted protocol for stranded RNA-seq [34].
- RNA Fragmentation & First-Strand Synthesis: RNA is first fragmented. First-strand cDNA synthesis is performed using random primers and reverse transcriptase [32] [33].
- Second-Strand Synthesis with dUTP Labeling: The second cDNA strand is synthesized using a nucleotide mix where dTTP is replaced with dUTP. This labels the second strand, distinguishing it from the first [32] [34].
- Adapter Ligation & Strand Degradation: Sequencing adapters are ligated to the double-stranded cDNA. Before PCR amplification, the second strand (containing uracils) is enzymatically degraded using Uracil-DNA-Glycosylase (UDG). This ensures only the first strand is amplified, preserving the strand information of the original RNA transcript [32] [34].
Key Technical Considerations for Robust Experiments
- RNA Quality is Paramount: RNA Integrity Number (RIN) >7 is generally recommended for high-quality sequencing. For degraded samples (RIN <6), ribosomal RNA (rRNA) depletion is more suitable than poly-A enrichment, which requires intact mRNA [35] [36].
- Minimize PCR Duplicates: The combination of low RNA input and high PCR cycle numbers can lead to a high rate of PCR duplicates, reducing library diversity and increasing noise. Using Unique Molecular Identifiers (UMIs) is highly recommended for low-input experiments to accurately identify and account for these artifacts [37].
- rRNA Depletion Strategy: While rRNA depletion (via probe-based hybridization or RNase H methods) enriches for informative transcripts, be aware of its variable efficiency and potential for off-target effects on some genes of interest [35].
The Scientist's Toolkit
| Category | Item | Function |
|---|---|---|
| Library Prep | dUTP Nucleotides | Labels the second strand of cDNA during synthesis, enabling its subsequent degradation and strand preservation [32] [34]. |
| Uracil-DNA-Glycosylase (UDG) | Enzymatically degrades the dUTP-labeled second cDNA strand, preventing its amplification [32] [34]. | |
| Strand-Switching Enzymes | Used in some protocols (not detailed in results) for cDNA synthesis and adapter addition. | |
| Sample QC | Agilent BioAnalyzer/TapeStation | Provides an electrophoretogram and RNA Integrity Number (RIN) to assess RNA quality [35] [36]. |
| Depletion/Enrichment | Oligo(dT) Magnetic Beads | Enriches for polyadenylated mRNA from high-quality, intact total RNA [35] [36]. |
| Ribosomal RNA Depletion Kits | Removes abundant rRNA using probes, allowing sequencing of non-polyadenylated transcripts and degraded RNA [35] [36]. | |
| Bias Mitigation | Unique Molecular Identifiers (UMIs) | Short random nucleotide tags added to each RNA molecule before amplification, allowing bioinformatic identification and removal of PCR duplicates [37]. |
Frequently Asked Questions & Troubleshooting
Q1: My RNA samples are partially degraded (RIN ~5). Can I still use a stranded protocol, and what is the best enrichment strategy? Yes, you can. For degraded RNA samples, ribosomal RNA depletion is strongly recommended over poly-A selection. Poly-A selection requires an intact 3' tail, which is often missing in degraded RNA. rRNA depletion targets ribosomal sequences directly and is not dependent on the RNA's integrity for enrichment [35] [36].
Q2: In my non-stranded data, I see evidence of expression in genomic regions with no annotated genes on that strand. What could this be? This is a classic limitation of non-stranded data. What appears to be expression from an unannotated region could, in fact, be antisense transcription originating from the opposite strand of an annotated gene. Without strand information, it is impossible to assign these reads correctly. Switching to a stranded protocol is essential for discovering and validating such antisense transcripts and long non-coding RNAs [32] [34].
Q3: My stranded library yield is low. What are the potential causes? Low yield in stranded preps can be attributed to its more complex workflow.
- Check RNA Input and Quality: Ensure you are using the recommended input amount and that the RNA is of high quality (RIN >7) [35] [36].
- Verify Enzymatic Steps: The additional enzymatic steps, particularly the second-strand degradation, can impact overall yield. Confirm that enzyme concentrations and reaction conditions are optimal [32].
- Assess PCR Amplification: An excessive number of PCR cycles can lead to duplicates and artifacts, but too few may result in low yield. Follow kit recommendations and consider using UMIs to monitor duplicate rates if you need to adjust cycles [37].
Q4: When is it acceptable to use the simpler, non-stranded protocol? Non-stranded RNA-seq is a valid and cost-effective choice when your primary goal is to quantify gene expression levels in an organism with a well-annotated genome, and you do not anticipate significant challenges from overlapping antisense transcripts. It is suitable for large-scale differential expression studies where strand information is not a priority [32] [33].
Decision Workflow for Protocol Selection
In the broader context of research on RNA-seq library preparation biases, the move towards low-input and single-cell RNA sequencing (scRNA-seq) has brought the issue of amplification bias into sharp focus. These advanced methods require significant amplification of minute starting amounts of genetic material, making them particularly susceptible to distortions that can compromise data integrity and lead to erroneous biological interpretations [2] [38]. This technical support center provides targeted troubleshooting guides and FAQs to help researchers identify, understand, and correct for these challenges, enabling more robust and reliable experimental outcomes.
Frequently Asked Questions (FAQs)
1. What are the primary sources of amplification bias in low-input RNA-seq? Amplification bias in low-input protocols primarily arises from the stochastic variation in amplification efficiency during the polymerase chain reaction (PCR). This can lead to the skewed representation of certain transcripts, where some molecules are over-amplified while others are under-amplified [2] [4]. Additional sources include the mispriming or nonspecific binding of primers [2], and the use of reverse transcriptases with varying efficiencies in converting RNA to cDNA, especially from fragmented or low-quality RNA templates [2] [38].
2. How does single-cell RNA-seq sensitivity relate to amplification bias? Sensitivity in scRNA-seq refers to the ability to detect a high number of transcripts per cell. Amplification bias directly threatens this sensitivity by introducing technical noise. Incomplete reverse transcription and uneven amplification of low-abundance transcripts can lead to "dropout" events, where a gene is falsely observed as not being expressed in a cell [38]. Therefore, mitigating amplification bias is crucial for improving sensitivity and accurately capturing a cell's true transcriptome.
3. What are the best practices to minimize amplification bias? Key strategies to minimize bias include:
- Using Unique Molecular Identifiers (UMIs): UMIs are random barcodes that label individual mRNA molecules before amplification. This allows bioinformatics tools to count original molecules and correct for amplification duplicates [38] [39].
- Optimizing PCR Cycles: Using the minimum number of PCR cycles necessary to generate sufficient library material is critical, as overcycling exacerbates biases [2] [4]. As shown in the table below, kits optimized for low input may require fewer cycles.
- Selecting Appropriate Enzymes: Using high-fidelity polymerases specifically designed for unbiased amplification of GC-rich regions can help reduce bias [2] [40].
- Employing Spike-In Controls: Adding known quantities of exogenous RNA transcripts can help monitor and normalize for technical variation, including amplification efficiency [38].
4. My sequencing data shows a high rate of duplicate reads. Is this related to amplification bias? Yes, a high duplication rate is a classic signature of over-amplification during library preparation [4]. When the starting material is limited, as in low-input and single-cell experiments, the same original molecules can be repeatedly sequenced after excessive PCR cycles. The use of UMIs is the most effective way to distinguish between technical duplicates (from PCR) and biological duplicates (from truly highly expressed genes) [39].
Troubleshooting Guides
Problem 1: Low Library Complexity and Sensitivity
Observation: The final sequencing data detects fewer genes than expected, with low unique molecule counts and a high degree of duplication.
| Possible Cause | Effect on Data | Suggested Solution |
|---|---|---|
| Low or degraded RNA input [2] [4] | Incomplete transcript coverage, high technical noise, and dropout of low-expression genes. | - Standardize cell lysis and RNA capture protocols [38].- Use high-quality, intact RNA with RIN > 8 for bulk low-input.- Assess RNA quality with a Bioanalyzer, not just spectrophotometry [4]. |
| Inefficient reverse transcription [2] | Loss of specific transcript classes and introduction of 3'-end bias. | - Use reverse transcriptases with high processivity and thermostability.- Incorporate template-switching oligonucleotides (TSO) for full-length cDNA capture [40]. |
| Suboptimal PCR amplification [2] [4] | Skewed representation of transcripts, over-representation of high-abundance genes, and high duplicate rates. | - Reduce the number of PCR cycles to the minimum necessary [2] [41].- Use polymerases known for uniform amplification across GC-content ranges (e.g., Kapa HiFi) [2].- For single-cell studies, always incorporate UMIs [38]. |
Problem 2: Amplification Bias and Uneven Coverage
Observation: Specific genes or regions are over-represented, coverage across transcript bodies is uneven, or there is a strong GC-content bias.
| Possible Cause | Effect on Data | Suggested Solution |
|---|---|---|
| PCR over-amplification [2] [41] | Formation of PCR artifacts (e.g., high-molecular-weight peaks on Bioanalyzer), increased chimeric reads, and flattening of expression distribution. | - Systematically titrate and reduce PCR cycle numbers. A guide from NEB suggests that extra high-molecular-weight peaks can be a direct result of over-amplification [41]. |
| Primer bias [2] | Under-representation of transcripts that do not prime efficiently with random hexamers. | - Use primers with balanced nucleotide representation.- For specific applications, consider ligation-based library methods that avoid random priming [2]. |
| Non-optimal polymerase | Preferential amplification of transcripts with neutral GC-content, leading to loss of AT-rich or GC-rich sequences. | - Switch to a polymerase mix engineered for unbiased amplification [2].- For extreme genomes, use PCR additives like TMAC or betaine [2]. |
The following table summarizes performance data from a comparison of different ultra-low-input RNA-seq kits, highlighting key metrics relevant to sensitivity and bias such as transcript detection and mappability.
Table 1: Sequencing Metrics Comparing Ultra-Low-Input cDNA Synthesis Methods (using 10 pg Mouse Brain Total RNA) [40]
| Metric | SMARTer Ultra Low v3 | SMART-Seq v4 | SMART-Seq2 Method |
|---|---|---|---|
| * cDNA Yield (ng)* | 4.7 - 6.0 | 10.6 - 12.6 | 8.1 - 11.2 |
| Number of Transcripts (FPKM >1) | ~9,400 | ~12,500 | ~10,100 |
| Reads Mapped to Genome (%) | 96 - 97% | 95 - 96% | 72 - 93% |
| Reads Mapped to Exons (%) | 73% | 76% | 66 - 67% |
| Key Improvement | Baseline | Higher sensitivity & yield | Incorporation of LNA technology |
Experimental Protocols
Protocol 1: Assessing Amplification Bias Using Spike-In Controls
Purpose: To quantitatively monitor technical variation and amplification efficiency across samples in a low-input RNA-seq experiment.
Materials:
- ERCC Spike-In Mix (or similar exogenous RNA control mix)
- Low-input RNA-seq library preparation kit (e.g., SMART-Seq v4)
- Bioanalyzer or TapeStation
- High-throughput sequencer
Methodology:
- Spike-In Addition: Prior to cDNA synthesis, add a defined, small amount of the ERCC spike-in RNA mix to each cell lysate or purified RNA sample. The amount should be consistent across all samples in the experiment [38].
- Library Preparation: Proceed with the standard low-input or single-cell library prep protocol, including reverse transcription, cDNA amplification, and library construction [40].
- Sequencing and Alignment: Sequence the libraries and align the reads to a combined reference genome that includes the host genome and the spike-in sequences.
- Data Analysis:
- Calculate the expression level (e.g., TPM or read counts) for each spike-in RNA.
- Plot the observed expression against the known input concentration for each spike-in. A protocol with low amplification bias will show a strong linear correlation across the entire dynamic range.
- Use the spike-in measurements to normalize the biological transcript data, correcting for global differences in amplification efficiency between samples [38].
Protocol 2: UMI-Based Correction for Amplification Duplicates
Purpose: To accurately count original mRNA molecules and remove technical duplicates introduced during PCR.
Materials:
- Library prep kit with integrated UMI design (e.g., 10x Genomics, STRT-seq)
- Computational tools for UMI deduplication (e.g., UMI-tools, zUMIs)
Methodology:
- Library Construction with UMIs: Use a protocol where UMIs are incorporated during the initial reverse transcription step, typically within the primers. This ensures each original cDNA molecule is tagged with a unique random barcode [39] [42].
- Sequencing: Perform paired-end sequencing. One read is dedicated to the transcript, and the other (or part of the same read) captures the UMI and cell barcode.
- Bioinformatic Deduplication:
- Extract UMIs and cell barcodes from the sequencing reads.
- Align the transcript reads to the reference genome.
- Group reads that align to the same genomic position and share the same cell barcode and UMI. These are considered PCR duplicates originating from a single mRNA molecule.
- Collapse these duplicate reads into a single count, representing one original molecule. This process yields a digital count of gene expression that is resistant to amplification bias [38] [42].
Workflow and Relationship Diagrams
Diagram 2: Amplification Bias Correction Strategies
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for Mitigating Bias in Low-Input RNA-seq
| Item | Function | Example Product/Technology |
|---|---|---|
| UMI-Adopted Kits | Enables accurate counting of original mRNA molecules by tagging each with a unique barcode before amplification, allowing for bioinformatic correction of PCR duplicates. | 10x Genomics Single Cell Kits, STRT-seq [38] [42] |
| High-Sensitivity RT Kits | Improves the efficiency of reverse transcription from very small amounts of RNA, often using template-switching technology to capture full-length transcripts. | SMART-Seq v4 Ultra Low Input RNA Kit [40] |
| Spike-In Control RNAs | Provides an external standard to monitor technical variance, detection limits, and amplification efficiency across experiments. | ERCC ExFold RNA Spike-In Mixes [38] |
| Bias-Reduced Polymerases | Polymerase enzymes engineered for uniform amplification efficiency across transcripts with varying GC-content, preventing skewed representation. | Kapa HiFi Polymerase [2] |
| Computational Tools | Software packages designed to perform UMI deduplication, batch effect correction, and normalization of sequencing data. | UMI-tools, NBGLM-LBC, Seurat, Harmony [38] [42] |
FAQs: Kit Selection and Performance
Q1: How do I choose the right RNA-seq kit based on my sample type and research goals?
The choice of kit critically depends on your sample's quality, quantity, and the RNA species you aim to capture. The following table summarizes the optimal applications for different kit technologies:
| Kit Technology / Brand | Ideal Sample Input | Key Applications and Strengths |
|---|---|---|
| SMARTer Ultra Low / SMART-Seq v4 [43] | 1–1,000 cells; 10 pg–10 ng total RNA (High quality, RIN ≥8) | Full-length transcript analysis for single-cells or ultra-low input; oligo(dT) priming for polyA+ mRNA [43]. |
| SMARTer Stranded [43] | 100 pg–100 ng total RNA | Maintains strand orientation (>99% accuracy); ideal for degraded RNA (e.g., FFPE) and non-polyadenylated RNA; requires rRNA depletion [43]. |
| SMARTer Universal Low Input [43] | 200 pg–10 ng total RNA (Degraded, RIN 2-3) | Random-primed for heavily degraded or non-polyadenylated RNA from sources like FFPE or LCM [43]. |
| NEBNext Ultra II Directional RNA [44] | Standard input ranges | Strand-specific library preparation; comprehensive troubleshooting guides for common library prep issues [44]. |
| Swift Biosciences Rapid RNA [45] | Not specified in results | Fast, stranded RNA-Seq library construction utilizing patented Adaptase technology [45]. |
Q2: What are the specific RNA quality requirements for these kits, and how should I assess them?
RNA Integrity Number (RIN) is a critical metric. SMARTer Ultra Low kits, which use oligo(dT) priming, require high-quality input RNA with a RIN ≥8 to ensure full-length cDNA synthesis [43]. In contrast, the SMARTer Universal Low Input Kit is designed for degraded RNA with a RIN of 2-3 [43]. For quantity and quality assessment, the use of the Agilent RNA 6000 Pico Kit is recommended, especially for low-concentration samples [43].
Q3: My RNA is degraded or from FFPE tissue. Which kit should I use?
For degraded samples, random-primed kits are superior to oligo(dT)-based ones. The SMARTer Stranded RNA-Seq Kit or the SMARTer Universal Low Input RNA Kit for Sequencing are specifically designed for this purpose [43]. These kits require prior ribosomal RNA (rRNA) depletion to prevent up to 90% of reads from mapping to rRNA [43].
Troubleshooting Guides
Troubleshooting Library QC and Sequencing Issues
Common problems observed during library quality control on the Bioanalyzer and their solutions are outlined below.
| Observation | Possible Cause | Suggested Solution |
|---|---|---|
| Bioanalyzer peak at 127 bp (Adapter-dimer) [44] | - Addition of undiluted adapter- RNA input too low- Inefficient ligation | - Dilute adapter (10-fold) before ligation- Perform a second PCR cleanup with 0.9X SPRI beads [44] |
| Bioanalyzer peaks below 85 bp (Primer dimers) [44] | - Incomplete removal of primers after PCR cleanup | - Clean up PCR reaction again with 0.9X AMPure beads [44] |
| High-molecular weight peak (~1,000 bp) [44] | - PCR over-amplification | - Reduce the number of PCR cycles [44] |
| Broad library size distribution [44] | - Under-fragmentation of RNA | - Increase RNA fragmentation time [44] |
| Low percentage of reads mapping to target after depletion [46] | - DNA contamination in input RNA- Compromised probe integrity- Incorrect target sequence used for probe design | - Treat sample with DNase I and purify- Verify probe integrity and storage conditions- Ensure the target sequence used for design is RNA, not cDNA [46] |
Troubleshooting Bias in Library Preparation
Library preparation is a major source of bias in RNA-seq data [2]. The table below details common biases and methods for improvement.
| Bias Source | Description | Suggestion for Improvement |
|---|---|---|
| Priming Bias [2] | Random hexamer priming can cause non-uniform read coverage. | For small RNA sequencing, use adapters with random nucleotides (degenerate bases) at ligation boundaries to mitigate sequence-dependent bias [47]. |
| Adapter Ligation Bias [2] [47] | T4 RNA ligases have substrate preferences, favoring certain RNA sequences over others. | Use adapters with random nucleotides at the extremities to be ligated to increase sequence diversity and ligation efficiency [2]. |
| PCR Amplification Bias [2] | Preferential amplification of cDNA with neutral GC content; bias propagates through cycles. | - Use polymerases like Kapa HiFi [2].- Reduce the number of PCR cycles [2].- For high GC content, use additives like TMAC or betaine [2]. |
| mRNA Enrichment Bias [2] | 3'-end capture bias during poly(A) selection. | For a broader transcriptome view, use rRNA depletion instead of poly-A enrichment to capture non-coding RNAs [2]. |
| Fragmentation Bias [2] | Non-random fragmentation using RNase III reduces library complexity. | Use chemical treatment (e.g., zinc) for fragmentation instead of enzymatic methods [2]. |
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function | Example / Note |
|---|---|---|
| Agilent RNA 6000 Pico Kit [43] | Accurately assesses RNA quantity and integrity (RIN) for low-concentration samples, a critical first step in sample QC. | Essential for quantifying ultra-low input and single-cell samples [43]. |
| Ribosomal RNA Depletion Kits [43] [46] | Removes abundant ribosomal RNA, dramatically increasing the percentage of informative mRNA and non-coding RNA reads. | Required for random-primed kits (e.g., SMARTer Stranded). Examples: RiboGone kit [43] or NEBNext RNA Depletion Core Reagent Set [46]. |
| NucleoSpin RNA XS Kit [43] | Purifies high-quality RNA from small sample sizes (e.g., up to 1x10^5 cultured cells) without a carrier. | Use of a poly(A) carrier is not recommended as it interferes with oligo(dT)-primed cDNA synthesis [43]. |
| SPRIselect / AMPure XP Beads [44] | Used for size-selective cleanup of DNA libraries, such as the removal of adapter dimers and primer artifacts. | A second cleanup (0.9X ratio) can resolve persistent adapter-dimer peaks [44]. |
| Degenerate Adapters [47] | Adapters with random nucleotides at ligation boundaries reduce sequence-specific ligation bias. | A key feature of kits like Bioo Scientific's NEXTflex V2 for small RNA sequencing, improving correlation with RT-qPCR data [47]. |
Experimental Workflow and Bias Mapping
RNA-seq Library Preparation Workflow
FAQs on RNA Quality and Degradation
What are the main challenges when working with RNA from FFPE samples?
Formalin-fixed paraffin-embedded (FFPE) tissues present several specific challenges for RNA extraction and sequencing. The formalin fixation process causes chemical modifications including RNA fragmentation, cross-linking of nucleic acids with proteins, and oxidation. The paraffin embedding process further degrades nucleic acids through heat and dehydration. Consequently, RNA from FFPE samples is typically highly fragmented and chemically modified, which leads to lower yields and can introduce biases in downstream applications like RNA-seq. This results in challenges such as lower sequencing coverage, potential loss of transcript diversity, and the introduction of sequencing artifacts. [48] [49] [50]
What quality metrics are most reliable for assessing FFPE RNA?
For FFPE-derived RNA, traditional metrics like the RNA Integrity Number (RIN) are often not adequate. Research supports the use of fragmentation-based metrics and PCR-based methods:
- DV200 (Percentage of RNA fragments >200 nucleotides) is a key indicator. While one Illumina report suggested a DV200 of 70% as a threshold, recent peer-reviewed studies indicate that a DV100 >80% provides the best indication of success for whole transcriptome sequencing, correlating well with gene detection rates. [50]
- PCR-based methods (qPCR or ddPCR) can quantify amplifiable RNA, which is crucial for determining effective input quantity for library prep, as fragmentation and cross-links can make a portion of the RNA unusable. [50]
- RQS (RNA Quality Score) is another parameter used by some analysers to assess integrity on a scale of 1 to 10. [48]
The table below summarizes key quality metrics and their interpretations for FFPE RNA:
Table 1: Quality Metrics for FFPE-Derived RNA
| Metric | Description | Interpretation / Recommended Threshold |
|---|---|---|
| DV200 | Percentage of RNA fragments >200 nucleotides | A common screening metric; higher values indicate less fragmentation. [48] |
| DV100 | Percentage of RNA fragments >100 nucleotides | >80% is a strong indicator of good gene diversity in sequencing. [50] |
| RIN | RNA Integrity Number | Less reliable for FFPE; often very low. Not recommended as a sole pass/fail criterion. [50] |
| PCR Amplification | Quantification of amplifiable RNA from a target gene | Informs required RNA input for library preparation; identifies samples with high levels of cross-linking. [50] |
| RNA Concentration | Measured by fluorometry (e.g., Qubit) | A minimum of 25 ng/μL is recommended for FFPE library prep. [51] |
How does RNA degradation impact transcript quantification in RNA-seq?
RNA degradation can significantly skew transcript quantification. The process is not uniform; different transcripts degrade at different rates, meaning degradation is non-random and can introduce bias. Studies show that even slight degradation (RIN ~6.7) can cause significant differences in the expression levels of many genes, particularly long non-coding RNAs (lncRNAs), compared to intact samples (RIN ~9.8). Principal Component Analysis (PCA) often shows that the level of degradation (RIN) can become the primary source of variation in the data, overwhelming biological signals. While standard data normalization is insufficient to correct for these effects, explicitly controlling for RIN or other degradation metrics in a linear model can recover a majority of the biological signal. [52] [53]
What are the minimum recommended RNA input values for successful FFPE RNA-seq?
Based on empirical data, the following pre-sequencing metrics are predictive of successful RNA-seq outcomes for FFPE samples:
Table 2: Pre-sequencing QC Recommendations for FFPE RNA-seq [51]
| Parameter | QC Pass Threshold | QC Fail Typical Value |
|---|---|---|
| Input RNA Concentration | ≥ 25 ng/μL | ~18.9 ng/μL |
| Pre-capture Library Qubit | ≥ 1.7 ng/μL | ~2.08 ng/μL |
| Post-sequencing Correlation | Spearman correlation ≥ 0.75 | < 0.75 |
A decision tree model using input RNA concentration and pre-capture library Qubit values can predict QC status with high accuracy (F-score of 0.848). [51]
Troubleshooting Guides
Problem: Low Library Yield from FFPE Samples
Potential Causes and Solutions:
- Cause: Poor RNA quality and high fragmentation.
- Solution: Prioritize samples with higher DV100/DV200 values. Use specialized RNA extraction kits designed for FFPE tissues, such as the Promega ReliaPrep FFPE Total RNA Miniprep or Roche kits, which have been shown to provide better quality and quantity. [48] Increase the input RNA quantity to compensate for the low amplifiable fraction. [50]
- Cause: Contaminants inhibiting enzymes.
- Solution: Re-purify the RNA using clean columns or beads. Ensure wash buffers are fresh and check purity via spectrophotometry (260/230 and 260/280 ratios). [4]
- Cause: Inefficient library preparation protocol.
- Solution: Use library prep kits with integrated DNA repair steps. For example, the NEBNext UltraShear FFPE DNA Library Prep Kit includes a repair mix that excises damaged bases and fills in overhangs, improving library conversion rates and data accuracy. [49] For RNA-seq, use rRNA depletion protocols (e.g., NEBNext rRNA Depletion) instead of poly(A) selection, which is less suitable for degraded RNA. [2] [51]
Problem: High Duplication Rates and Biased Coverage
Potential Causes and Solutions:
- Cause: Over-amplification during library prep.
- Cause: Random hexamer priming bias.
- Solution: Be aware that random hexamers can bind with different efficiencies, causing uneven coverage. Some bioinformatics tools offer read count reweighing schemes to adjust for this bias. [2]
- Cause: Low input RNA leading to loss of library complexity.
Problem: High Adapter Dimer Formation
Potential Causes and Solutions:
- Cause: Suboptimal adapter-to-insert molar ratio.
- Solution: Titrate the adapter concentration. Too much adapter promotes adapter-dimer formation, while too little reduces ligation yield. [4]
- Cause: Inefficient purification after ligation.
- Solution: Optimize bead-based cleanups. Use the correct bead-to-sample ratio to exclude small fragments like adapter dimers. Avoid over-drying the bead pellet, which leads to inefficient resuspension and sample loss. [4]
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for FFPE and Degraded RNA Workflows
| Item | Function | Example Products / Methods |
|---|---|---|
| FFPE RNA Extraction Kits | Specialized lysis buffers and protocols to reverse cross-links and recover fragmented RNA. | Promega ReliaPrep FFPE Total RNA Miniprep, Roche High Pure FFPE RNA Isolation Kit [48] |
| RNA Quality Assessment | Analyze fragmentation and quantify amplifiable RNA to determine sequencing input. | Agilent Bioanalyzer (DV200, DV100), Qubit Fluorometer, qPCR/ddPCR assays [48] [51] [50] |
| rRNA Depletion Kits | Remove abundant ribosomal RNA to enrich for mRNA and other transcripts, preferable for degraded RNA. | NEBNext rRNA Depletion Kit (Human/Mouse/Rat) [51] |
| Library Prep with Repair Enzymes | Enzymatic mixes that repair DNA damage (nicks, gaps, deaminated bases) common in FFPE samples. | NEBNext UltraShear FFPE DNA Library Prep Kit [49] |
| Robust Polymerases | Enzymes that reduce amplification bias, especially for GC-rich or difficult templates. | Kapa HiFi Polymerase [2] |
Experimental Workflows and Visualization
FFPE RNA Quality Assessment and Sequencing Workflow
The following diagram outlines a recommended workflow for processing FFPE tissues from sample to sequencing, incorporating key quality control checkpoints.
Impact of RNA Degradation on Data Analysis
This diagram illustrates the logical relationship between RNA degradation and its effects on sequencing data and interpretation.
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference in the data generated by 3' mRNA-seq and whole transcriptome sequencing?
The core difference lies in the distribution of sequencing reads along the transcript. Whole Transcriptome Sequencing (WTS) generates reads uniformly across the entire length of all transcripts, enabling the detection of splice variants, fusion genes, and novel isoforms [54]. In contrast, 3' mRNA-seq generates reads preferentially from the 3' end of transcripts, providing a digital count-like output ideal for gene expression quantification but without isoform-level information [54] [55].
Table: Fundamental Data Characteristics
| Feature | Whole Transcriptome Sequencing | 3' mRNA-Seq |
|---|---|---|
| Read Distribution | Uniform coverage across the entire transcript body [55] | Strong bias towards the 3' end of genes [55] |
| Quantification Basis | Proportional to transcript length and abundance [55] | Directly proportional to transcript count (one read per transcript) [54] [55] |
| Detection Sensitivity | Detects more differentially expressed genes (DEGs), especially longer transcripts [54] [55] | Detects more short transcripts at lower sequencing depths; fewer overall DEGs [54] [55] |
| Isoform Resolution | Yes (alternative splicing, novel isoforms) [54] | No [54] [56] |
Q2: For a large-scale drug screening project requiring high throughput and low cost, which method is preferable?
3' mRNA-seq is the strongly recommended choice. Methods like BRB-seq and DRUG-seq are designed for ultra-high-throughput, using early sample barcoding and multiplexing to process up to 384 samples simultaneously [57] [58]. This drastically reduces hands-on time and library preparation cost, which can be up to 25 times cheaper than standard RNA-seq [58]. Furthermore, the significantly lower sequencing depth required (1-5 million reads/sample) further reduces overall project costs [54] [58].
Q3: My research requires the detection of alternative splicing events. Which protocol should I use?
You must use Whole Transcriptome Sequencing. 3' mRNA-seq methods, by design, sequence only the 3' end of transcripts and therefore cannot provide information on alternative splicing, novel isoforms, or fusion genes [54] [56]. Only WTS provides uniform coverage along the entire transcript, allowing for the identification of different splice variants and the investigation of complex transcriptional events [54].
Q4: How should I prepare libraries if my RNA samples are degraded (e.g., from FFPE tissues)?
3' mRNA-seq is often more robust for degraded samples. Because it exclusively sequences the 3' end of transcripts, it is less affected by the 5'-to-3' degradation that commonly occurs in FFPE and other challenging sample types [54] [58]. The protocol's reliance on the 3' region, which is often better preserved, allows for successful gene expression profiling even when RNA Integrity Number (RIN) values are low (<6) [58].
Troubleshooting Guides
Problem: Low Library Yield or Failed Library Preparation
Low yield can occur at multiple steps. The table below outlines common causes and solutions.
Table: Troubleshooting Low Library Yield
| Symptoms | Potential Root Cause | Corrective Action |
|---|---|---|
| Low yield starting from purified RNA. | Input RNA is degraded or contaminated with salts, phenol, or other inhibitors [4]. | Re-purify input RNA; verify quality using fluorometric methods (e.g., Qubit) and check 260/230 and 260/280 ratios [4]. |
| Adapter-dimer peaks in Bioanalyzer traces. | Inefficient ligation or tagmentation; suboptimal adapter-to-insert molar ratio [4]. | Titrate adapter concentrations; ensure fresh enzyme and optimal reaction conditions [4]. |
| Low complexity libraries, high duplication rates. | Overly aggressive purification leading to sample loss; too many PCR cycles [4]. | Optimize bead-based cleanup ratios; avoid over-drying beads; reduce the number of PCR amplification cycles [4]. |
| Inconsistent failures across technicians. | Pipetting errors and deviations from the protocol [4]. | Use master mixes to reduce pipetting; implement detailed SOPs with highlighted critical steps; introduce technician checklists [4]. |
Problem: Discrepancy in Gene Detection Between 3' mRNA-seq and Whole Transcriptome Data
This is an expected methodological difference, not a technical failure.
- Cause 1: Transcript Length Bias. WTS assigns more reads to longer transcripts, while 3' mRNA-seq is largely insensitive to transcript length [55]. Therefore, WTS will have higher power to detect differential expression in longer genes.
- Solution: Be aware of this inherent bias during data interpretation. The biological conclusions at the pathway level are typically highly consistent between the two methods, even if the specific lists of DEGs differ [54].
- Cause 2: Poor 3' Annotation. The performance of 3' mRNA-seq is highly dependent on accurate genome annotation of transcript 3' ends [54].
- Solution: For model organisms, ensure you are using the most recent genome annotation. For non-model organisms, it may be necessary to improve the 3' annotation, as insufficient annotation will lead to reduced mapping rates [54].
Experimental Protocols & Workflows
Detailed Protocol: 3' mRNA-seq (BRB-seq)
The following workflow is adapted from the highly multiplexed BRB-seq protocol [57] [58].
Key Steps Explained:
- Reverse Transcription with Barcoding: Total RNA is used as input. Reverse transcription is initiated using barcoded oligo(dT) primers. These primers contain a sample-specific barcode and a Unique Molecular Identifier (UMI), which labels each individual mRNA molecule [57] [58].
- Early Sample Pooling: After reverse transcription, all uniquely barcoded samples are pooled into a single tube. This is a critical step that enables massive multiplexing and significant reduction in reagent use and hands-on time [57].
- Second-Strand Synthesis & Tagmentation: The pooled cDNA undergoes second-strand synthesis. The resulting double-stranded cDNA is then fragmented and tagged with sequencing adapters in a single step using Tn5 transposase (tagmentation) [58].
- Library Amplification: PCR is performed with indexed primers to add full sequencing adapters and further amplify the library, which is then ready for sequencing [57].
Detailed Protocol: Full-Length RNA-seq (Traditional Workflow)
This describes a standard whole transcriptome approach, such as the Illumina TruSeq stranded mRNA protocol [59].
Key Steps Explained:
- mRNA Enrichment: Total RNA is processed to enrich for mRNA, typically via poly(A) selection to capture polyadenylated transcripts, or through ribosomal RNA (rRNA) depletion to also retain non-polyadenylated RNAs [54] [59].
- RNA Fragmentation: The enriched mRNA is randomly fragmented into shorter pieces [55].
- cDNA Synthesis and Adapter Ligation: The fragmented RNA is reverse transcribed into cDNA using random primers, ensuring coverage across the entire transcript. This is followed by second-strand synthesis. Adapters are then ligated to the ends of the double-stranded cDNA fragments [59].
- Library Amplification: The adapter-ligated fragments are PCR-amplified with indexed primers to create the final sequencing library [59].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Reagents for RNA-seq Library Construction
| Reagent / Enzyme | Function in Protocol | Key Consideration |
|---|---|---|
| Barcoded Oligo(dT) Primers | Initiates reverse transcription from the poly(A) tail and labels cDNA with sample barcode and UMI [57] [58]. | Crucial for early multiplexing in 3' mRNA-seq. UMI allows for accurate digital counting by correcting for PCR duplicates. |
| Tn5 Transposase | Simultaneously fragments double-stranded cDNA and ligates sequencing adapters (tagmentation) [60] [58]. | Significantly streamlines workflow. Can be produced in-house for major cost reduction [60]. |
| Template Switching Oligo (TSO) | Used in some full-length protocols (e.g., SMART-seq) to allow reverse transcriptase to add universal sequences to the 5' end of cDNA [61]. | Can introduce artifacts via "strand invasion"; newer methods avoid it [62]. |
| Ribonuclease Inhibitor | Protects RNA templates from degradation during reverse transcription and library prep [60]. | Essential for maintaining RNA integrity, especially in low-input or long protocols. |
| M-MuLV Reverse Transcriptase | Synthesizes first-strand cDNA from an RNA template [60] [61]. | Variants with high processivity and terminal transferase activity are used for template-switching protocols [61]. |
From Problems to Solutions: Mitigating Bias Through Experimental Design and Protocol Refinement
The reliability of RNA sequencing (RNA-seq) data is fundamentally dependent on the quality of the input RNA. Unlike DNA, RNA is a highly labile molecule that can be easily degraded by ubiquitous RNases, or compromised by factors such as heat, contaminated chemicals, and inadequate buffer conditions [63]. Since the ultimate quality of sequencing data is largely dependent on the starting material, evaluating RNA integrity is a critical pre-analytical step to ensure experimental success and reproducibility [63] [64].
Within the broader context of research on RNA-seq library preparation biases, the integrity of the input RNA is a primary source of pre-analytical variation. Degraded RNA can lead to multiple technical biases, including altered gene expression measurements, reduced library complexity, and spurious results in downstream analyses [2]. This guide provides a structured framework for assessing input RNA quality, focusing on the RNA Integrity Number (RIN) and other key metrics, to help researchers mitigate these risks and generate robust, reliable data.
Understanding and Implementing the RNA Integrity Number (RIN)
What is the RNA Integrity Number (RIN)?
The RNA Integrity Number (RIN) is a standardized algorithm developed by Agilent Technologies to assign an integrity value to RNA samples. It is a numerical value on a scale of 1 to 10, where 10 represents completely intact RNA and 1 represents fully degraded RNA [63] [65]. The RIN algorithm was developed to replace the traditional and highly subjective method of judging RNA quality by the 28S-to-18S ribosomal RNA ratio on agarose gels [65]. By utilizing capillary electrophoresis and a proprietary Bayesian learning model, RIN provides a more objective, reproducible, and automated assessment of RNA integrity [63] [66] [65].
How is RIN Calculated?
The RIN algorithm analyzes the entire electrophoretic trace of an RNA sample, not just the ribosomal peaks. Key features used in the calculation include [65]:
- The total RNA ratio: The ratio of the area under the 18S and 28S rRNA peaks to the total area under the electropherogram.
- The height of the 28S peak: This peak is typically degraded more quickly than the 18S peak.
- The "fast region": The area between the 18S and 5S rRNA peaks, which contains degradation products.
- The marker region: The presence of small, degraded RNA fragments near the lower marker.
The following diagram illustrates the logical workflow and key features analyzed by the RIN algorithm.
Interpreting RIN Scores for Downstream Applications
A general guideline is that a RIN score of 7 to 10 is considered acceptable for most downstream applications [63]. However, different molecular techniques have varying sensitivity to RNA degradation. The table below summarizes the recommended RIN thresholds for common applications.
Table 1: Acceptable RIN Score Ranges for Common Applications
| Application | Recommended RIN Score | Rationale |
|---|---|---|
| RNA Sequencing (RNA-seq) | 8 - 10 [63] | Ensures full-length transcript coverage and minimizes 3'/5' bias. |
| Microarray Analysis | 7 - 10 [63] | High integrity is needed for accurate probe hybridization. |
| qPCR | >7 [63] | Specific short amplicons can be targeted, but high quality is still preferred. |
| RT-qPCR | 5 - 6 [63] | Can often be performed on more degraded RNA by designing amplicons near the 3' end. |
| Gene Arrays | 6 - 8 [63] | Moderate integrity may be sufficient depending on the platform. |
Frequently Asked Questions (FAQs) and Troubleshooting Guides
FAQ: Common Questions on RIN and RNA QC
Q1: My sample has a low RIN. Can I still use it for RNA-seq? Proceeding with a low RIN sample (e.g., <7) for standard RNA-seq protocols is not advisable, as it can lead to severe 3' bias and inaccurate gene expression quantification [2]. However, if the sample is irreplaceable, consider using specialized library prep kits designed for degraded RNA (e.g., those emphasizing 3' sequencing) and increase the sequencing depth. Be aware that data interpretation will be limited.
Q2: My RIN is over 8, but my RNA-seq data still shows strong 3' bias. Why? A high RIN indicates good starting RNA integrity. However, 3' bias can also be introduced during library preparation, particularly by protocols that use oligo-dT primers for cDNA synthesis or poly-A enrichment [67]. This bias can be exacerbated by subtle RNA degradation or suboptimal reaction conditions. Check your library prep protocol and ensure all steps are performed on ice with RNase-free reagents.
Q3: Is RIN applicable to all sample types, including prokaryotic RNA? The standard RIN algorithm was developed and validated for eukaryotic RNA, where the dominant species are 18S and 28S rRNAs. For prokaryotic samples, which have 16S and 23S rRNAs, the algorithm is different and may be less validated [65]. Furthermore, RIN is often unsuitable for plants or samples with mixed eukaryotic-prokaryotic content, as it cannot differentiate between the ribosomal RNAs from different kingdoms [65].
Q4: What are the alternatives to RIN for quality assessment? A common alternative metric is the DV200 (Percentage of RNA fragments > 200 nucleotides), which is often considered more reliable for highly degraded samples, such as those from Formalin-Fixed Paraffin-Embedded (FFPE) tissues [66]. Post-sequencing QC metrics, such as Gene Body Coverage and the percentage of reads mapping to exons, are also critical for validating RNA quality after data generation [68] [64].
Troubleshooting Guide: Common RNA Quality Issues
Table 2: Troubleshooting Guide for RNA Quality Issues
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Low RIN Score / RNA Degradation | - RNase contamination during isolation.- Delays in sample processing or freeze-thaw cycles.- Improper storage conditions. | - Use fresh RNase inhibitors and dedicated RNase-free reagents/consumables [63].- Flash-freeze tissues in liquid nitrogen immediately after collection [2].- Minimize freeze-thaw cycles by aliquoting RNA [2]. |
| Low RNA Concentration | - Insufficient starting material.- Inefficient extraction from certain tissue types.- Sample loss during bead cleanups. | - Use high-concentration RNA extraction kits. For single-cells, use specialized ultra-low input kits [69].- Ensure proper homogenization of tissues.- Use a strong magnetic stand and follow bead drying times precisely to prevent loss [69]. |
| High rRNA Contamination in Seq Data | - Inefficient ribosomal RNA depletion during library prep. | - Optimize rRNA depletion protocols. Use probe-based depletion kits for higher efficiency [2] [64]. |
| Genomic DNA Contamination | - Inefficient DNase I treatment during RNA extraction. | - Perform an on-column DNase I digestion. For persistent contamination, a secondary DNase treatment can be added, which has been shown to significantly reduce intergenic reads [70]. |
| Inconsistent RIN Scores | - Variable sample collection or processing times.- Use of different operators or reagent batches. | - Standardize the time from collection to freezing across all samples [71].- Implement batch control by processing samples from different experimental groups together to minimize batch effects [71] [64]. |
The Scientist's Toolkit: Essential Reagents and Equipment
Table 3: Essential Research Reagents and Equipment for RNA QC
| Item | Function/Benefit | | :--- :--- | | Agilent Bioanalyzer 2100 or 4200 TapeStation | Automated electrophoresis systems that generate digital electropherograms and automatically calculate the RIN [66] [65]. | | RNA Extraction Kits (e.g., Qiagen RNeasy, mirVana) | Silica-gel-based column procedures for high-yield, high-quality RNA isolation. The mirVana kit is noted for superior performance with non-coding RNAs and low concentrations [2]. | | RNase Inhibitors | Reagents added to lysis and elution buffers to inactivate RNases and preserve RNA integrity during extraction [63]. | | DNase I, RNase-free | Enzyme used to digest and remove contaminating genomic DNA during RNA purification, crucial for accurate RNA-seq results [70]. | | PAXgene Blood RNA Tubes | Specialized collection tubes for blood samples that immediately stabilize RNA, preserving its in vivo gene expression profile [70]. | | SMART-Seq Single-Cell Kits | Designed for cDNA synthesis and library prep from ultra-low input RNA (e.g., single cells), which is highly sensitive to degradation and loss [69]. |
Best Practices and Detailed Methodologies
A Comprehensive RNA QC Workflow
Implementing a rigorous, multi-layered QC strategy is essential for successful RNA-seq. The following workflow diagram outlines the key checkpoints from sample collection to sequencing.
Experimental Protocol: Assessing RNA Integrity Using the Agilent Bioanalyzer
This protocol provides a generalized methodology for assessing RNA quality, a critical step before library preparation.
Objective: To determine the concentration and integrity (RIN) of total RNA samples using the Agilent Bioanalyzer 2100.
Materials and Reagents:
- Agilent RNA 6000 Nano Kit (contains RNA Nano chips, gel matrix, dye, ladder, and reagents)
- The RNA samples to be tested
- RNase-free water and pipette tips
- Thermal cycler or heat block
- Vortex mixer and spin centrifuge
- Agilent 2100 Bioanalyzer instrument
Methodology:
- Chip Preparation: Place an RNA Nano chip on the chip priming station. Pipette 9 µL of the gel matrix into the well marked with a black circle.
- Prime Chip: Close the priming station and press the plunger until it is held by the clip. Wait for exactly 30 seconds, then release the clip. Wait a further 5 seconds before pulling the plunger back to its start position.
- Load Gel: Pipette 9 µL of the gel matrix into the two other wells marked with a white "G".
- Prepare Ladder and Samples:
- Ladder: Pipette 5 µL of the RNA 6000 Nano marker into the ladder well. Then, pipette 1 µL of the RNA ladder into the same well.
- Samples: Pipette 5 µL of the RNA 6000 Nano marker into each sample well. Then, pipette 1 µL of each RNA sample into separate sample wells.
- Run Chip: Place the chip into the Agilent 2100 Bioanalyzer adapter and vortex for 1 minute at 2400 rpm. Insert the adapter into the instrument and start the run using the associated software.
- Data Analysis: The software will automatically generate an electropherogram, calculate the RIN for each sample, and provide the RNA concentration.
Key Considerations:
- Ensure all reagents are at room temperature before starting.
- Always include the ladder for proper sizing and RIN calibration.
- The recommended RNA concentration for accurate RIN calculation is greater than 50 ng/µL; concentrations below 25 ng/µL are not recommended [63].
- For samples with low concentration but high integrity, the DV200 metric can be a useful supplementary measure [66].
Frequently Asked Questions
1. What are the primary sources of bias in rRNA depletion protocols? The main sources of bias include off-target depletion, where probes hybridize to and remove non-rRNA transcripts of interest, and technical variability in the depletion efficiency itself [35]. The degree of bias can depend on the specific depletion method; for instance, some protocols show more reproducible off-target effects, while others exhibit greater variability between experiments [35].
2. My sequencing data shows low coverage of mRNA after rRNA depletion. What could be the cause? This is often a result of inefficient rRNA removal, meaning too much of your sequencing capacity is still being consumed by ribosomal reads [18]. This can be caused by using a depletion kit that is not optimized for your specific organism [72], using degraded reagents, or following a suboptimal protocol. Ensure your method is species-specific and that you are using high-quality, fresh reagents [4].
3. How does the choice between rRNA depletion and polyA enrichment affect my results? The choice fundamentally determines which part of the transcriptome you can analyze.
- PolyA Enrichment: Selects for messenger RNAs (mRNAs) with polyadenylated tails. It is cost-effective but introduces a 3' end bias and excludes all non-polyadenylated RNAs, such as many long non-coding RNAs (lncRNAs) and histone genes [35] [72] [73].
- rRNA Depletion: Removes ribosomal RNA, thereby enriching for all other RNA species, including both polyA+ and non-polyA transcripts. This is crucial for studying total RNA, non-coding RNAs, and samples with degraded RNA (like FFPE), but requires careful optimization to minimize off-target effects [35] [72].
4. Can I use a depletion kit designed for one species on a different organism? This is not recommended and is a common source of poor depletion efficiency. Ribosomal RNA sequences, while somewhat conserved, can have unique structures and sequences in different organisms [72]. For example, kits designed for humans/mice/rats often perform poorly on Drosophila melanogaster due to fragmentation of its 28S rRNA [72]. Always use a depletion method validated for your specific research organism.
Troubleshooting Guide: Common Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High rRNA reads post-depletion | Inefficient hybridization, wrong probe set for species, degraded RNase H (in enzymatic methods), low probe:RNA ratio [72] [74]. | Verify organism compatibility of kit/probes. Use DOE to optimize probe & bead ratios. Use fresh enzyme aliquots [74]. |
| Loss of specific mRNAs | Off-target hybridization of depletion probes to non-rRNA transcripts [35]. | Check literature for known off-targets. Validate findings with an alternative method (e.g., qPCR). Switch or re-design probe sets. |
| Low library yield | Overly aggressive purification post-depletion; sample loss during multiple clean-up steps [4]. | Optimize bead-based clean-up ratios to minimize loss. Avoid over-drying magnetic beads. |
| High variability between replicates | Inconsistent handling during hybridization or capture steps; reagent degradation [35] [4]. | Standardize incubation times and temperatures. Use master mixes for reagents. Implement rigorous quality control of input RNA. |
Comparison of Ribosomal RNA Depletion Methods
The following table summarizes the key characteristics, advantages, and limitations of common rRNA depletion strategies, based on comparative studies.
| Depletion Method | Principle | rRNA Removal Efficiency | Pros | Cons & Off-Target Risks |
|---|---|---|---|---|
| Biotinylated Probe Hybridization & Pull-down [18] | Biotinylated DNA probes bind rRNA; complexes removed with streptavidin beads. | High (e.g., comparable to former RiboZero) [18]. | High efficiency; physically removes rRNA. | Risk of off-target pull-down; can be variable [35]. |
| RNase H-mediated Depletion [35] [72] | DNA probes hybridize to rRNA; RNase H enzyme degrades RNA-DNA hybrids. | ~97% reported for optimized, species-specific protocols [72]. | No physical separation step; less sample loss. | RNase H can have non-specific activity, digesting off-target hybrids [18]. |
| CRISPR-DASH (Post-library) [73] | Cas9 nuclease cleaves rRNA sequences in cDNA library using specific sgRNAs. | Highly effective for targeted rRNA genes [73]. | Minimal off-target effects; operates on amplified cDNA. | Requires specialized sgRNA design and construction. |
| Commercial Pan-Prokaryotic Kits [18] | Pre-designed probes for a range of bacteria (e.g., RiboMinus, MICROBExpress). | Variable; may not target 5S rRNA [18]. | Convenience. | Lower efficiency if not perfectly matched to species [18]. |
Experimental Protocol: RNase H-based Depletion
This protocol outlines a cost-effective and efficient method for ribosomal RNA depletion using RNase H, which can be tailored to your organism of interest [72].
1. Probe Design
- Objective: Design single-stranded DNA oligos (~60-100 nt) that are reverse-complementary to the 18S, 28S, and 5S rRNA sequences of your target organism.
- Critical Step: For high specificity, perform a BLAST analysis to ensure probes do not have significant homology to non-rRNA mRNA or ncRNA sequences. This minimizes off-target depletion [72].
2. Hybridization
- Combine 100 ng - 1 µg of total RNA with the pool of DNA probes in a hybridization buffer.
- Thermal Cycling:
- Denature at 95°C for 2 minutes.
- Hybridize at 55-65°C for 10-30 minutes. This temperature is critical for specific probe binding and should be optimized [74].
3. Enzymatic Digestion
- Add RNase H enzyme and its corresponding buffer to the hybridization mix.
- Incubate at 37°C for 30 minutes to digest the RNA-DNA hybrids.
4. RNA Clean-up
- Use a standard RNA clean-up kit (e.g., magnetic beads) to remove the DNA probes and degraded rRNA fragments.
- Elute the depleted RNA, which is now enriched for non-ribosomal transcripts and ready for library construction.
The workflow for this protocol is summarized in the following diagram:
The Scientist's Toolkit: Essential Reagents for rRNA Depletion
| Item | Function | Example Use-Case |
|---|---|---|
| Species-Specific DNA Probes | Hybridize to complementary rRNA sequences for targeted depletion. | Core component of in-house RNase H or pull-down protocols; essential for non-model organisms [72] [18]. |
| RNase H Enzyme | Enzymatically degrades the RNA strand in RNA-DNA hybrids. | Used in RNase H-based depletion methods after probe hybridization [72]. |
| Streptavidin Magnetic Beads | Bind to biotinylated probes for physical removal of rRNA complexes. | Used in probe pull-down methods (e.g., riboPOOLs, in-house protocols) [18] [73]. |
| RNA Clean-up Kits | Purify RNA after depletion, removing enzymes, salts, and probes. | Essential post-depletion step before library preparation [4]. |
| Design of Experiments (DOE) | A statistical framework to efficiently optimize multiple protocol factors. | Used to simultaneously optimize probe concentration, bead amount, and incubation time to maximize efficiency and minimize cost [74]. |
A Framework for Systematic Protocol Optimization
Troubleshooting a protocol by changing one variable at a time is inefficient. The statistical Design of Experiments (DOE) framework allows you to explore multiple factors and their interactions simultaneously [74].
Application to rRNA Depletion: A study used DOE to optimize an rRNA depletion protocol by testing three key factors:
- Antisense rRNA probe level
- Amount of total RNA input
- Amount of streptavidin beads
The analysis revealed significant interactions between these factors, leading to an optimized protocol that removed more rRNA while using fewer reagents and at a lower cost, all with only 36 experimental runs [74]. The logic of this approach is illustrated below.
FAQs: Understanding and Diagnosing Batch Effects
What are batch effects and why are they a critical concern in large-scale omics studies?
Batch effects are technical variations in data that are unrelated to the biological objectives of a study. They are introduced due to variations in experimental conditions over time, using data from different labs or machines, or different analysis pipelines [75]. In large-scale studies, their impact is profound: they can introduce noise that dilutes biological signals, reduce statistical power, and lead to misleading, biased, or non-reproducible results [75]. In the worst cases, they are a paramount factor contributing to the irreproducibility of scientific findings, which can result in retracted articles and invalidated research [75]. For example, in a clinical trial, a change in RNA-extraction solution led to an incorrect risk calculation for 162 patients, 28 of whom subsequently received incorrect chemotherapy regimens [75].
At which stages of my RNA-seq experiment are batch effects most likely to be introduced?
Batch effects can emerge at virtually every step of a high-throughput study [75]. The table below summarizes common sources.
Table 1: Common Sources of Batch Effects in RNA-seq Experiments
| Stage | Specific Source | Description of Bias |
|---|---|---|
| Sample Preparation & Storage | Sample Storage Conditions [75] | Variations in storage temperature, duration, or number of freeze-thaw cycles. |
| RNA Extraction [2] | Different methods (e.g., TRIzol vs. column-based) can cause selective loss of certain RNA species. | |
| Input RNA [2] | Low quantity or quality (degraded) input RNA can introduce strong biases. | |
| Library Construction | mRNA Enrichment [2] | Poly(A) enrichment can cause 3'-end capture bias, under-representing transcripts or parts of transcripts. |
| Fragmentation [2] | Non-random fragmentation (e.g., using RNase III) reduces library complexity. | |
| Primer Bias [2] | Random hexamer primers can bind non-randomly, leading to mispriming and uneven coverage. | |
| Adapter Ligation [8] | T4 RNA ligases have sequence-dependent preferences, over-representing fragments that co-fold with adapters. | |
| PCR Amplification [2] | Stochastically and preferentially amplifies different cDNA molecules, a major source of bias. | |
| Sequencing | Sequencing Platform [2] | Different platforms (e.g., Illumina, Ion Torrent) have inherent biases in base calling and error profiles. |
My study involves samples processed over many months. How can I design the experiment to minimize batch effects?
A carefully considered experimental setup is your first and most powerful defense against batch effects [76].
- Plate Layout: When planning multi-plate experiments, arrange your samples so that experimental conditions and controls are distributed across all plates and processing batches. This prevents "confounding," where a biological group of interest is processed entirely in a single batch [76].
- Randomization: Process samples in a randomized order rather than grouping all samples from one condition together.
- Reference Materials: Incorporate a common reference material (a well-characterized control sample) into every batch you run. This allows for robust batch-effect correction later by scaling study sample data relative to the reference [77].
- Replicates: Always include a sufficient number of biological replicates (independent biological samples per condition) to account for natural variation. While 3 is typical, between 4-8 per group is recommended for greater reliability [76].
Troubleshooting Guides: Solutions for Common Scenarios
I have already collected data from a confounded study design (e.g., all "Control" samples were sequenced in Batch 1, all "Treatment" in Batch 2). Can this data be salvaged?
This is a challenging but common scenario. When biological groups are completely confounded with batch, it becomes nearly impossible to distinguish true biological differences from technical batch variations [77]. Most standard batch-effect correction algorithms (BECAs) fail or may remove the biological signal of interest in this situation [77].
Solution: The most effective solution is to leverage reference materials. If you included a common reference sample (e.g., a commercial RNA reference or a pooled sample) in both Batch 1 and Batch 2, you can use a ratio-based correction method [77]. This method transforms the absolute expression values of your study samples into ratios relative to the values of the reference sample measured in the same batch. This scaling effectively cancels out the batch-specific technical variation, allowing for a valid comparison [77]. Without reference samples, the options for reliable analysis are severely limited.
My RNA-seq data shows clear batch-driven clustering in PCA plots. What are my options for computational correction?
A plethora of batch-effect correction algorithms (BECAs) exist. The choice of method depends on your data type and the level of confounding. The following workflow outlines a general decision-making process for selecting and applying a BECA.
Table 2: Comparison of Common Batch Effect Correction Algorithms (BECAs)
| Method Name | Category | Applicable Data Types | Key Principle | Considerations |
|---|---|---|---|---|
| ComBat / ComBat-seq / ComBat-ref [78] [77] | Non-procedural (Model-based) | Bulk RNA-seq (Count data) | Uses a parametric empirical Bayes framework to adjust for batch effects. ComBat-ref adjusts batches towards a low-dispersion reference batch. | Can be powerful but risks over-correction if batch and biology are confounded. ComBat-ref shows superior sensitivity/specificity [78]. |
| Ratio-based (Ratio-G) [77] | Reference-based | Multi-omics (Transcriptomics, Proteomics, Metabolomics) | Scales absolute feature values of study samples relative to those of a concurrently profiled reference material. | Highly effective, especially in confounded designs. Requires prior planning to include reference samples in every batch [77]. |
| Harmony [77] [79] | Procedural (Iterative) | scRNA-seq, Bulk RNA-seq | Iteratively corrects PCA embeddings to align batches while preserving biological variation. | Does not return a corrected expression matrix, but a corrected embedding for clustering/visualization [79]. |
| Seurat v3 [79] | Procedural (Anchoring) | scRNA-seq | Identifies "anchors" (mutual nearest neighbors) between batches to correct the data. | Effective for integrating diverse single-cell datasets. A widely used standard. |
| Order-Preserving Methods [79] | Procedural (Deep Learning) | scRNA-seq | Uses monotonic deep learning networks to correct effects while preserving the original rank-order of gene expression within a cell. | Maintains inter-gene correlations and differential expression patterns, improving biological interpretability [79]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Batch Effect Management
| Reagent / Material | Function in Managing Batch Effects |
|---|---|
| Reference Materials (e.g., Quartet Project references) [77] | Provides a universally available, well-characterized standard to be run in every experimental batch. Enables ratio-based correction and cross-batch quality control. |
| Spike-in Controls (e.g., SIRVs) [76] | Artificial RNA sequences added to each sample in known quantities. They act as an internal standard for normalization, helping to quantify technical variability and assess dynamic range. |
| Standardized RNA Extraction Kits | Using the same kit and, ideally, the same lot of reagents across the entire study minimizes variability introduced during RNA isolation [2]. |
| Bias-Reducing Enzymes (e.g., Kapa HiFi Polymerase, trRnl2 K227Q) [2] [8] | High-fidelity PCR polymerases reduce GC-content bias during amplification. Engineered ligases like trRnl2 K227Q reduce sequence-dependent bias during adapter ligation [8]. |
| rRNA Depletion Kits | An alternative to poly(A) selection for mRNA enrichment, which can help avoid 3'-end capture bias, especially for degraded samples (e.g., FFPE) [2]. |
A foundational challenge in RNA sequencing is the preparation of high-quality libraries from minimal amounts of starting RNA, a common scenario when working with rare cell populations or limited clinical samples such as formalin-fixed paraffin-embedded (FFPE) tissues [2] [80]. The core of this challenge lies in a critical balancing act: applying sufficient PCR amplification to generate a sequenceable library without allowing this process to distort the true biological representation of transcripts within the sample. Amplification bias, where certain cDNA molecules are amplified more efficiently than others, can propagate through the experiment, compromising data integrity and leading to erroneous biological conclusions [2]. This technical issue forms a significant focus within modern research on RNA-seq biases, driving the development of improved laboratory protocols and bioinformatic corrections. The following guide provides targeted troubleshooting and strategic advice to help researchers navigate these low-input challenges effectively.
Troubleshooting Guide: Low-Input RNA-seq Libraries
When library yield or quality is unsatisfactory, a systematic approach to troubleshooting is essential. The table below outlines common symptoms, their potential causes, and recommended corrective actions.
Table 1: Troubleshooting Guide for Low-Input RNA-seq Experiments
| Observed Problem | Potential Root Cause | Corrective Action & Solutions |
|---|---|---|
| Low final library yield [4] | Poor input RNA quality or contaminants inhibiting enzymes [4]. | Re-purify input sample; use fluorometric quantification (e.g., Qubit) over absorbance; ensure high purity (260/230 > 1.8) [4]. |
| Inefficient adapter ligation [4]. | Titrate adapter-to-insert molar ratios; ensure fresh ligase and optimal reaction conditions [4]. | |
| Overly aggressive purification or size selection [4]. | Optimize bead-to-sample ratios during clean-up steps to minimize loss of desired fragments [4]. | |
| High duplication rates [81] | Excessive PCR amplification from low starting material [2] [81]. | Reduce the number of PCR cycles; use polymerases designed for high fidelity (e.g., Kapa HiFi) [2]. Incorporate Unique Molecular Identifiers (UMIs) to bioinformatically correct for PCR duplicates [20]. |
| Skewed gene expression / High bias | Preferential amplification of certain transcripts (e.g., based on GC content) [2]. | For AT/GC-rich targets, use PCR additives like TMAC or betaine [2]. Validate with ERCC spike-in controls to assess technical performance [20]. |
| Primer bias during reverse transcription or PCR [2]. | Use random priming during cDNA synthesis instead of oligo-dT for degraded samples [2]. | |
| High ribosomal RNA (rRNA) content | Inefficient rRNA depletion, especially problematic with low-input total RNA [80] [81]. | Select kits with proven enzymatic rRNA depletion methods [80] [81]. For blood samples, ensure protocols also deplete globin mRNA [20]. |
Optimized Experimental Protocols for Low-Input RNA
Strategic Selection of Library Preparation Kits
The choice of library preparation kit is pivotal for the success of low-input RNA-seq studies. Recent comparative studies evaluate kits based on their input requirements and performance with challenging samples.
Table 2: Comparison of Library Prep Kit Performance Characteristics
| Kit / Workflow Name | Low-Input Performance | Key Advantages & Application Notes |
|---|---|---|
| TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 (Kit A) [80] | Requires 20-fold less RNA input than comparator (Kit B) while achieving comparable gene expression quantification [80]. | Ideal for extremely limited samples, albeit potentially requiring increased sequencing depth to compensate for higher duplication rates [80]. |
| Illumina Stranded Total RNA Prep with Ribo-Zero Plus (Kit B) [80] | Standard input requirements; generates high-quality data. | Demonstrates superior alignment performance, lower rRNA content, and lower duplication rates with sufficient input [80]. |
| Watchmaker RNA Library Prep with Polaris Depletion [81] | Optimized for a range of sample types, including FFPE and whole blood. | Significantly reduces duplication rates, improves rRNA and globin depletion, and detects 30% more genes compared to standard methods [81]. |
Recommended Step-by-Step Protocol Modifications
- RNA Input and Quality Control: For samples with low integrity (e.g., FFPE), use a high sample input if possible [2]. Assess RNA quality using the DV200 metric (percentage of fragments > 200 nucleotides); samples with DV200 > 30% are generally usable for RNA-seq, though higher values are preferable [80].
- rRNA Depletion and cDNA Synthesis: Employ robust rRNA depletion methods (e.g., enzymatic removal) rather than poly-A selection for degraded samples or when analyzing non-polyadenylated RNAs [2] [20]. Use random primers instead of oligo-dT for reverse transcription to mitigate 3'-bias, which is exacerbated in low-quality RNA [2].
- Library Amplification and Cleanup: Use the minimum number of PCR cycles necessary to obtain sufficient library yield [2] [4]. Consider using polymerases known for uniform amplification across different transcript sequences [2]. Perform careful size selection to exclude adapter dimers and retain the desired fragment range, optimizing bead-based clean-up ratios to prevent loss of material [4].
Visualizing the Low-Input RNA-seq Optimization Strategy
The following diagram illustrates the primary decision points and optimization strategies for a successful low-input RNA-seq experiment, helping to balance amplification and representation.
Diagram 1: Low-input RNA-seq optimization workflow.
Frequently Asked Questions (FAQs)
Q1: What is the minimum amount of RNA required for a standard RNA-seq library? While standard protocols often recommend 100 ng to 1 μg of total RNA [82], specialized kits have been successfully demonstrated with inputs as low as 1 ng, and ultra-low input protocols can work with even less [80] [20]. The required input depends on RNA quality, the kit used, and the desired sequencing depth.
Q2: How can I accurately quantify gene expression when I've had to heavily amplify my library? The most effective method is to incorporate Unique Molecular Identifiers (UMIs) during library preparation [20]. UMIs are short random barcodes added to each original cDNA molecule before amplification. After sequencing, bioinformatic tools can identify and collapse reads with identical UMIs, correcting for both PCR duplication bias and errors, thereby restoring accurate quantitative representation [20].
Q3: My FFPE RNA is degraded. Should I use poly-A selection or rRNA depletion? For degraded FFPE samples, rRNA depletion is strongly recommended [20]. Poly-A selection relies on intact 3' poly-adenylated tails, which are often compromised in degraded RNA. Depletion methods remove ribosomal RNA regardless of transcript integrity, allowing for a more comprehensive and representative profile of the remaining RNA fragments [2] [20].
Q4: How many PCR cycles are too many during library amplification? There is no universal number, but the goal is always to use the minimum number of cycles necessary to obtain sufficient library for sequencing [4]. Excessive cycles manifest as high duplication rates and can introduce significant amplification bias [2] [4]. Monitoring library complexity and yield at each step helps determine the optimal cycle number for a given protocol and input amount.
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Reagents for Mitigating Low-Input and Amplification Biases
| Reagent / Tool | Primary Function | Role in Bias Mitigation |
|---|---|---|
| UMIs (Unique Molecular Identifiers) [20] | Molecular barcoding of original cDNA molecules. | Enables bioinformatic correction for PCR duplication bias and errors, ensuring accurate transcript quantification. |
| ERCC Spike-In Controls [20] | Exogenous synthetic RNA mixes of known concentration. | Allows for standardization and assessment of technical performance, including sensitivity, dynamic range, and accuracy of the entire workflow. |
| High-Fidelity Polymerase (e.g., Kapa HiFi) [2] | Amplification of cDNA libraries. | Reduces preferential amplification of specific sequences, leading to more uniform coverage across transcripts with varying GC content. |
| rRNA Depletion Probes (e.g., Ribo-Zero, Polaris) [80] [81] | Selective removal of ribosomal RNA. | Increases the proportion of informative reads mapping to the transcriptome, improving sequencing efficiency and gene detection, crucial for low-input and degraded samples. |
| Random Primers [2] | Initiation of reverse transcription. | Prevents 3'-end bias caused by oligo-dT priming, which is especially important when working with partially degraded RNA (e.g., from FFPE). |
FAQs: Core Concepts and Application
Q1: What are the primary differences between ERCC and SIRV spike-in controls, and when should I use each?
ERCC and SIRV controls are designed for distinct but complementary purposes. The ERCC (External RNA Controls Consortium) spike-in mix consists of 92 synthetic, single-isoform transcripts that span a wide, known concentration range (over six orders of magnitude) [83]. They are primarily used to assess the sensitivity, dynamic range, and accuracy of RNA-seq experiments [84] [83]. In contrast, the SIRV (Spike-in RNA Variants) controls are a family of modules engineered to mimic the complexity of eukaryotic transcriptomes, including features like alternative splicing, alternative transcription start and end sites, and overlapping genes [84] [85]. A key SIRV module contains 69 isoforms derived from 7 artificial genes [86]. SIRVs are therefore essential for validating experiments focused on transcript isoform detection and quantification [84]. Your choice depends on the experimental goal: use ERCCs to evaluate dynamic range and limit of detection, and SIRVs to benchmark performance in splice variant analysis and other complex transcriptome features [84] [87] [86].
Q2: How do I determine the correct amount of spike-in RNA to add to my sample?
A general rule of thumb is to spike in an amount such that approximately 1% of your total sequencing reads map to the spike-in genome (the "SIRVome" or ERCC references) [84] [85]. For a standard bulk RNA-seq experiment starting with 100 ng of total RNA, this typically translates to about 50 picograms of spike-in RNA, given that mRNA represents roughly 5% of total RNA [85]. For specific applications like single-cell RNA-seq, where total RNA per cell is much lower (e.g., 20 pg), the amount must be drastically reduced—for example, to the 200 femtogram range [85]. The exact amount should be empirically determined and tailored to your specific RNA fraction (total, ribosomal depleted, or poly(A)-enriched) and the expected RNA content of your sample [84]. Lexogen provides an "Experiment Designer" tool to help calculate recommended spike-in ratios based on your experimental parameters [85].
Q3: Can spike-in controls be used for normalization in differential expression analysis, and what are the potential pitfalls?
Yes, spike-in controls can be used for normalization, particularly in scenarios where global transcriptional changes are expected, such as when transcription factors are knocked down [88] [89]. Standard gene-based normalization methods (e.g., the median ratio method in DESeq2) assume that most genes are not differentially expressed, an assumption violated in such experiments. In these cases, using spike-ins provides a robust external reference [89].
However, potential pitfalls exist. The accuracy of normalization depends on the precise and consistent addition of the same amount of spike-in RNA to each sample and the assumption that spike-in and endogenous transcripts are affected similarly by technical biases [89]. Inconsistent spike-in addition or divergent behavior can compromise normalization. Research indicates that in plate-based single-cell protocols, the variance in added spike-in volume is quantitatively negligible, supporting its reliability for scaling normalization [89]. If you encounter issues, such as an unexpectedly low number of differentially expressed genes after spike-in normalization in DESeq2, you can try the type='iterate' option in the estimateSizeFactors function or consider using dedicated packages like RUVSeq [88].
Q4: My spike-in controls are not being detected after sequencing. What could have gone wrong?
Several points in the experimental workflow could lead to this issue:
- Spike-in Addition: The spike-in mix may have been added at too low a concentration, forgotten, or degraded due to improper storage or handling.
- Library Preparation: The spike-in RNAs are polyadenylated, making them compatible with poly(A)-enrichment protocols [84]. If a protocol that does not target poly(A)+ RNA is used (e.g., total RNA without enrichment), the spike-ins will not be captured. Ensure your library prep method is compatible.
- Data Analysis: During read alignment, you must map reads to a combined reference that includes both the endogenous genome (e.g., human, mouse) and the spike-in genomes (ERCC and/or SIRV) [84]. Failure to provide a combined reference genome and annotation file (.gtf) at the alignment step will result in no reads mapping to the spike-ins [88].
Troubleshooting Common Experimental Issues
Problem: Inconsistent Normalization Results with Spike-ins
Symptoms: After using spike-ins for normalization (e.g., in DESeq2), the number of differentially expressed genes is unexpectedly low or sample clustering (PCA, heatmaps) appears worse than with standard normalization [88].
Solutions:
- Verify Spike-in Performance: Check that the spike-in controls themselves are behaving as expected. Plot the measured log-fold changes of the spike-in transcripts against their expected values. A strong correlation indicates the spike-ins are performing well [88].
- Iterative Normalization: In DESeq2, try using the iterative method for estimating size factors by setting
type='iterate'in theestimateSizeFactorsfunction. This method can be more robust in some situations [88]. - Alternative Methods: Consider using the
RUVSeqpackage, which is designed to use control genes (like spike-ins) to remove unwanted variation and often integrates well with DESeq2 [88]. - Re-evaluate Necessity: In many cases, especially without global transcriptional shifts, standard median-ratio normalization is sufficient and robust. Spike-in normalization is most critical when there are massive, genome-wide changes in mRNA content [88] [89].
Problem: Low or Variable Spike-in Read Counts Across Samples
Symptoms: The percentage of reads mapping to spike-ins varies dramatically between samples in the same experiment, complicating normalization.
Solutions:
- Standardize Addition: Ensure the spike-in mix is thoroughly vortexed and centrifuged before use. Use calibrated pipettes and consistent technique when adding the mix to each sample. Using a master mix for the spike-ins can improve consistency.
- Check Sample Quality and Quantity: Variable spike-in counts can reflect true biological differences in the endogenous RNA content of your samples. For instance, if one sample has significantly more endogenous RNA than another but the same amount of spike-in is added, the proportion of spike-in reads will be lower in that sample [84]. This is not an error but a feature—the spike-ins are correctly reporting this disparity for proper normalization.
- Optimize Dilution: For sensitive applications like single-cell RNA-seq, ensure the spike-in stock is diluted accurately and added at an appropriate step in the protocol (e.g., to the lysis buffer) [85].
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Key Commercially Available Spike-in Control Sets
| Product Name | Key Components | Primary Application | Key Features |
|---|---|---|---|
| ERCC Spike-in Mixes [83] | 92 single-isoform RNAs | Assessing sensitivity, dynamic range, and technical performance. | Concentrations span 6 orders of magnitude. |
| SIRV-Set 2 (Isoform Mix E0) [86] | 69 isoform transcripts at equimolar concentration. | Validating isoform detection and quantification workflows. | All transcripts are at the same concentration, ideal for testing detection bias. |
| SIRV-Set 3 (Isoform E0 & ERCC) [86] | 69 SIRV isoforms + 92 ERCC RNAs. | Comprehensive quality assessment covering both isoform complexity and abundance dynamic range. | Combines the strengths of both systems in one mix. |
| SIRV-Set 4 (Complete Module) [86] | 69 SIRV isoforms + 92 ERCCs + 15 long SIRVs (4-12 kb). | Full workflow validation, especially for protocols handling long transcripts. | Adds a length dimension (up to 12 kb) to the complexity and abundance controls. |
Table 2: Summary of Spike-in Control Applications and Properties
| Control Type | Ideal for Normalization? | Best Used For | Considerations |
|---|---|---|---|
| ERCC | Yes, especially with global transcriptional changes [88] [89]. | Establishing limit of detection, dynamic range, and linearity [83]. | Single-isoform, does not address splice variant complexity [84]. |
| SIRV (Isoform Module) | Primarily for QC, not routine normalization [85]. | Benchmarking splice variant analysis, identifying pipeline biases in isoform assignment [84] [87]. | Available in different mixes (E0, E1, E2) to model different expression hierarchies [86]. |
| Combined SIRV+ERCC | Potentially more powerful, but more complex. | Holistic pipeline validation from abundance to isoform discovery [86]. | Requires more sequencing reads; analysis must separate the two components. |
Experimental Protocols and Workflows
Detailed Protocol: Incorporating Spike-in Controls in RNA-seq
Materials:
- Purified RNA sample or cell lysate.
- Selected spike-in mix (e.g., SIRV-Set 3 or ERCC).
- RNA-seq library preparation kit.
Method:
- Spike-in Addition:
- Thaw the spike-in mix on ice and briefly centrifuge.
- Calculate the required volume of spike-in mix to achieve your target percentage (e.g., ~1% of mRNA reads) based on your sample's estimated RNA content [84] [85].
- Critical Step: Add the calculated volume of spike-in mix directly to your sample before any RNA purification or library preparation steps. This can be done to purified RNA or at an upstream stage like cell lysis or homogenization, ensuring the spikes undergo the entire wet-lab procedure alongside the endogenous RNA [84].
Library Preparation and Sequencing:
- Proceed with your standard RNA-seq library prep protocol (e.g., poly(A) enrichment, ribosomal RNA depletion, etc.). The SIRV and ERCC RNAs are polyadenylated, making them compatible with poly(A)+ selection protocols [84].
- The spike-in controls are compatible with all major NGS platforms, including Illumina, Ion Torrent, PacBio, and Oxford Nanopore Technologies [84].
- Sequence the libraries as planned.
Data Analysis:
- Reference Generation: Create a combined reference for read alignment. This involves concatenating the endogenous reference genome (e.g., GRCh38) with the spike-in sequence files (FASTA for the "SIRVome" and/or ERCCs). Similarly, combine the corresponding annotation files (.gtf) [84] [88].
- Read Mapping: Map the sequencing reads to this combined reference using your preferred aligner (e.g., STAR, HISAT2).
- Quality Control and Normalization:
- Separate the read counts for endogenous genes and spike-in transcripts.
- Use the spike-in counts for quality assessment, calculating metrics like coefficient of deviation, accuracy, and precision by comparing observed coverage to expected values [84].
- For normalization in differential expression tools like DESeq2, use the
controlGenesoption in theestimateSizeFactorsfunction to specify the spike-in transcripts [88].
The following diagram illustrates the core experimental workflow.
Logical Framework for Troubleshooting Spike-in Normalization
The following decision tree helps diagnose common problems with spike-in normalization.
Benchmarking Performance: Quantitative Framework for Assessing Library Prep Quality
Within the broader investigation of RNA-seq library preparation biases, rigorous quality control stands as a fundamental pillar for generating reliable and interpretable data. The highly complex workflow of RNA-seq, from sample preservation to sequencing, is susceptible to numerous technical variations that can introduce significant bias, potentially compromising downstream biological interpretations [2]. This guide focuses on three essential technical metrics—rRNA retention, duplication rates, and mapping efficiency—that serve as critical indicators of library quality and experimental soundness. By providing troubleshooting guidelines and best practices, we aim to empower researchers to diagnose, rectify, and prevent common issues, thereby enhancing the fidelity of their transcriptomic studies.
FAQ: Understanding the Core Metrics
What are rRNA retention, duplication rate, and mapping efficiency, and why are they important?
These three metrics provide a snapshot of the technical success of your RNA-seq library preparation and sequencing run.
- rRNA Retention: This measures the percentage of sequencing reads that originate from ribosomal RNA (rRNA). In eukaryotic cells, rRNA can constitute 80-98% of total RNA, yet it is typically uninformative for gene expression studies [90]. A high rRNA retention rate wastes sequencing resources, reducing the depth for meaningful targets like messenger RNA.
- Duplication Rate: This is the percentage of reads that are perfect duplicates, aligning to the exact same genomic start and end positions. While some duplication is expected for highly expressed genes, an abnormally high rate often indicates low library complexity, potentially stemming from insufficient input RNA, over-amplification during PCR, or other biases that limit the diversity of fragments sequenced [91] [4].
- Mapping Efficiency (or Mapping Rate): This crucial metric indicates the percentage of total sequenced reads that successfully align to the reference genome or transcriptome [90]. A low mapping rate can signal issues like sample contamination, poor RNA quality, the presence of adapter sequences, or an incomplete reference [92].
What are the typical acceptable ranges for these metrics?
Acceptable ranges can vary based on the organism, library preparation method, and experimental goals. The following table summarizes general benchmarks for high-quality data.
Table 1: Benchmark Ranges for Key RNA-seq Quality Metrics
| Metric | Excellent | Acceptable | Cause for Concern | Primary Influence |
|---|---|---|---|---|
| rRNA Retention | < 5% | 5% - 10% | > 10% [90] | RNA enrichment/depletion method [91] |
| Duplication Rate | < 20% | 20% - 50% | > 50% [91] | Library complexity, PCR amplification [4] |
| Mapping Efficiency | > 80% | 70% - 80% | < 70% [93] [92] | RNA integrity, contamination, reference quality |
How does library preparation method specifically impact rRNA retention?
The choice between poly(A) selection and ribodepletion is the most significant factor influencing rRNA retention.
- Poly(A) Selection: This method uses oligo(dT) beads to capture polyadenylated mRNA, effectively excluding non-polyA RNAs like rRNA. It performs best with high-quality, intact RNA but will fail to capture non-polyadenylated transcripts and is unsuitable for degraded samples (e.g., FFPE) or prokaryotic RNA [91] [93].
- Ribodepletion: This method uses probes to remove rRNA sequences, leaving behind other RNA species, including both coding and non-coding RNA. It is applicable to degraded samples and prokaryotes but may be less efficient than poly(A) selection. A study comparing methods found that RNase H-based ribodepletion could achieve remarkably low rRNA retention (0.1%), while other methods showed higher levels (e.g., Ribo-Zero at 11.3%) [91].
Troubleshooting Guides
High rRNA Retention
- Problem: A high percentage of reads align to ribosomal RNA.
- Symptoms: Low percentage of informative (e.g., exonic) reads; reduced power to detect rare transcripts.
- Root Causes & Solutions:
- Cause: Inefficient rRNA depletion.
- Solution: For prokaryotic samples or degraded RNA, ensure ribodepletion is used instead of poly(A) selection. Titrate the depletion probes to ensure optimal performance [2].
- Cause: Degraded RNA.
- Cause: Inefficient rRNA depletion.
Diagram: Troubleshooting workflow for high rRNA retention.
High Duplication Rate
- Problem: An abnormally high fraction of PCR duplicate reads.
- Symptoms: Low library complexity; skewed representation of transcript abundance; potential masking of true biological variation.
- Root Causes & Solutions:
- Cause: Over-amplification during library PCR.
- Cause: Insufficient input RNA.
- Cause: Inefficient fragmentation or ligation.
- Solution: Optimize fragmentation parameters (time, energy, enzyme concentration) and ensure correct adapter-to-insert molar ratios to maximize library diversity [4].
Table 2: Troubleshooting High Duplication Rates
| Root Cause | Diagnostic Check | Corrective Action |
|---|---|---|
| Over-amplification | Review library prep protocol for high PCR cycle number. | Reduce the number of PCR cycles; use high-fidelity polymerases [2]. |
| Low Input RNA | Check BioAnalyzer/Qubit data for low starting yield. | Increase input RNA; use low-input/amplification protocols (e.g., SMART, NuGEN) [91]. |
| Fragmentation/Ligation | Check electropherogram for unexpected fragment size or adapter-dimer peaks. | Optimize fragmentation; titrate adapter:insert ratio [4]. |
Low Mapping Efficiency
- Problem: A low percentage of reads successfully align to the reference.
- Symptoms: High percentage of unmapped reads; potential for false negatives in expression analysis.
- Root Causes & Solutions:
- Cause: Ribosomal RNA reads not aligning.
- Solution: rRNA sequences are often multi-copy and may be filtered out by aligners as multi-mapping reads. Check the aligner's log for a high count of multi-mapping reads. Consider using a reference that includes all rRNA genomic loci [92].
- Cause: Adapter contamination or poor raw read quality.
- Solution: Perform rigorous quality control on raw reads. Trim adapter sequences and low-quality bases using tools like Trimmomatic or FASTX-Toolkit before alignment [93].
- Cause: Sample contamination or use of an incorrect reference.
- Cause: Ribosomal RNA reads not aligning.
Diagram: Troubleshooting workflow for low mapping efficiency.
The Scientist's Toolkit: Research Reagent Solutions
Selecting the appropriate library preparation kit and reagents is critical for optimizing the quality metrics discussed. The following table outlines common solutions and their functions.
Table 3: Key Reagents and Methods for RNA-seq Library Preparation
| Reagent / Method | Function / Principle | Impact on Key Metrics |
|---|---|---|
| Poly(A) Selection | Enriches for polyadenylated mRNA using oligo(dT) beads. | Minimizes rRNA retention. Best for intact, eukaryotic RNA [93]. |
| Ribodepletion (e.g., Ribo-Zero, RNase H) | Uses probes or enzymes to remove ribosomal RNA. | Reduces rRNA retention. Essential for degraded RNA, prokaryotes, and non-polyA transcripts [91] [93]. |
| RNase H Method | A specific ribodepletion method using RNase H to degrade rRNA. | Can achieve exceptionally low rRNA retention (e.g., 0.1%), effective for low-quality RNA [91]. |
| SMART / NuGEN (Ovation) | Protocols designed for low-quantity input RNA, often using template-switching. | Maintains library complexity, helping to control duplication rates in low-input scenarios [91]. |
| Kapa HiFi Polymerase | A high-fidelity PCR polymerase used in library amplification. | Reduces amplification bias and over-duplication during the PCR step [2]. |
| ERCC Spike-in Controls | Synthetic RNA molecules added to the sample in known concentrations. | Acts as an external standard to assess technical performance, including accuracy of quantification and detection of bias [95]. |
The selection of RNA-seq library preparation kits represents a critical methodological decision that directly influences the composition of differentially expressed gene (DEG) lists and subsequent biological interpretations. Within the broader context of RNA-seq library preparation biases research, understanding how technical choices propagate through analytical workflows is essential for experimental reproducibility. This technical support center resource addresses how kit selection introduces variability in DEG detection and provides troubleshooting guidance for researchers seeking to optimize their transcriptomic studies.
How Library Preparation Choices Affect Differential Expression Results
Experimental Evidence: Quantitative Impacts of Technical Choices
Recent research has systematically quantified how library preparation decisions affect DEG detection and functional analysis outcomes:
Table 1: Impact of Sequencing Strategy on DEG Concordance
| Experimental Factor | Effect on Read Mapping | Impact on DEG Lists | Functional Enrichment Concordance |
|---|---|---|---|
| Single-end (SE) vs. Paired-end (PE) | 3.3-9.4% reduction in uniquely assigned reads with SE; 20% increase in multimapped reads with SE | 5% false positives and 5% false negatives with SE compared to PE | ~40% discordance in top GO terms with SE vs. PE |
| Non-stranded (NS) vs. Stranded Protocol | 116% average increase in ambiguous reads with NS approach | Additional 1-2% increase in false positives/negatives with NS | Striking differences in top GO terms with NS |
| Statistical Approach | Not applicable | Significant variation in protein lists from same datasets | Lower consistency when varying biological relevance criteria |
Research demonstrates that using single-end reads produces DEG lists containing approximately 5% false positives and 5% false negatives compared to paired-end reads [96]. The non-stranded approach further compounds these errors, increasing false positives and negatives by an additional 1-2 percentage points [96]. These technical differences substantially impact downstream biological interpretation, with functional enrichment analysis showing as little as 40% concordance in top Gene Ontology terms between single-end and paired-end approaches [96].
Troubleshooting Guide: Addressing Common Technical Challenges
Table 2: Library Preparation Issues and Solutions
| Problem Category | Failure Signals | Root Causes | Corrective Actions |
|---|---|---|---|
| Sample Input & Quality | Low library yield; smear in electropherogram; low complexity | Degraded RNA; contaminants (phenol, salts); inaccurate quantification | Re-purify input; use fluorometric quantification (Qubit); check 260/230 ratios (>1.8) |
| Adapter Contamination | Sharp ~70-90 bp peaks in BioAnalyzer; high adapter dimer signals | Improper adapter-to-insert molar ratio; inefficient ligation | Titrate adapter:insert ratios; optimize ligation conditions; use dual-size selection |
| Amplification Bias | Overamplification artifacts; high duplicate rate; GC bias | Too many PCR cycles; inefficient polymerase; primer exhaustion | Reduce PCR cycles; use high-fidelity polymerase; employ PCR additives for GC-rich regions |
| Mapping & Quantification Issues | Low uniquely assigned reads; high multimapped or ambiguous reads | Non-stranded protocol; short read length; repetitive regions | Switch to strand-specific protocol; use paired-end reads; increase read length |
The most problematic library preparation step is typically PCR amplification, which stochastically introduces biases that propagate through subsequent analysis [2]. Overcycling during amplification introduces size bias, increases duplicate rates, and flattens expression distributions [4]. For low-input samples, these effects are exacerbated, potentially leading to significant distortions in DEG lists [2].
Frequently Asked Questions
How much does strand-specificity affect functional enrichment results?
Strand-specific protocols significantly improve the reliability of functional enrichment results. Studies show striking differences in the top Gene Ontology terms when comparing stranded versus non-stranded approaches, with as little as 40% concordance in significantly enriched terms [96]. The non-stranded protocol generates a 116% average increase in ambiguous reads, where the genomic location is known but the read could belong to multiple features on different strands [96]. This ambiguity leads to misassignment of reads to incorrect genes, which in turn affects the DEG list and subsequent pathway analysis.
Can normalization methods compensate for library preparation biases?
Normalization methods can mitigate some technical variability but cannot fully compensate for fundamental library preparation biases. Methods like TMM (Trimmed Mean of M-values) assume most genes are not differentially expressed, which becomes problematic when global shifts in expression occur or when a few highly expressed genes dominate the transcriptome [97]. Between-sample normalization corrects for sequencing depth differences, but cannot resolve issues like strand-specific read misassignment or adapter contamination [98] [97]. The normalization approach itself introduces assumptions that can affect downstream results, with Bullard et al. finding that normalization procedure had greater impact on differential expression results than the choice of test statistic [97].
When might single-end sequencing be acceptable despite its limitations?
Single-end sequencing may be an acceptable trade-off when sequencing budget constraints would otherwise prevent adequate biological replication [96]. While SE reads produce DEG lists with approximately 5% false positives and false negatives compared to PE reads, the cost savings can be redirected toward increasing biological replicates, thereby improving statistical power [96]. Research indicates that when used in association with gene set enrichment analysis (GSEA), single-end reads can generate biologically accurate conclusions despite the higher error rate in individual DEG calls [96].
Research Reagent Solutions
Table 3: Essential Reagents for Minimizing Technical Bias
| Reagent Category | Specific Examples | Function | Bias Reduction |
|---|---|---|---|
| Stranded Library Prep Kits | Illumina TruSeq Stranded mRNA; Illumina Stranded Total RNA | Maintain strand orientation during cDNA synthesis | Reduces ambiguous read mapping by 116% compared to non-stranded protocols |
| High-Fidelity Polymerases | Kapa HiFi Polymerase | PCR amplification with minimal bias | Reduces preferential amplification of GC-neutral fragments |
| RNA Preservation Reagents | mirVana miRNA isolation kit; Non-cross-linking organic fixatives | Maintain RNA integrity and yield | Minimizes degradation artifacts; improves yield for low-abundance transcripts |
| Bead-Based Cleanup | AMPure XP beads | Size selection and purification | Reduces adapter dimer contamination; improves library complexity |
| RNA Quality Assessment | Agilent Bioanalyzer/TapeStation; Fragment Analyzer | Assess RNA Integrity Number (RIN) | Identifies degraded samples before library preparation |
Experimental Workflow and Concordance Relationships
Technical choices in RNA-seq library preparation, particularly strand-specificity and read type, significantly impact the concordance of differential expression results and subsequent biological interpretation. Researchers must weigh the trade-offs between cost and data fidelity when designing transcriptomic studies. Adopting stranded protocols and paired-end sequencing maximizes reliability, while single-end approaches may be acceptable when coupled with increased biological replication and gene set enrichment analysis. Consistent reporting of library preparation methodologies is essential for experimental reproducibility and meaningful cross-study comparisons.
Frequently Asked Questions
How do technical biases specifically affect the reproducibility of pathway enrichment results? Technical biases during RNA-seq library preparation can systematically skew gene expression data. This is a significant contributor to the broader "reproducibility crisis" in biomedical research [99]. When these technical factors, such as sample quality, are imbalanced between the disease and control groups in a study, they act as confounding variables [100]. This imbalance can lead to the false identification of hundreds of differential genes and the overrepresentation of stress-response pathways, ultimately resulting in biologically irrelevant pathway enrichment results that fail to validate in subsequent studies [100].
What are the most common sources of bias in RNA-seq library prep that I should watch for? The most common issues can be categorized as follows [4] [2]:
- Sample Quality: Using degraded RNA or samples with contaminants (e.g., from FFPE preservation) inhibits enzymes and reduces library complexity [4] [2].
- Fragmentation & Ligation: Non-random RNA fragmentation and inefficient adapter ligation can introduce sequence-length biases and create adapter-dimer contaminants [4] [2].
- Amplification (PCR): Too many PCR cycles preferentially amplifies certain transcripts, leading to high duplication rates and skewed representation [4] [2].
- Priming Bias: The use of random hexamers during reverse transcription can cause uneven coverage across transcripts [2].
My pathway analysis shows enrichment for common stress-response pathways. Could this be a technical artifact? Yes, this is a classic red flag. Studies have identified thousands of genes that can serve as "quality markers," and their presence is often associated with sample stress [100]. An enrichment analysis of these markers frequently highlights transcription factors and miRNAs related to stress response. If your dataset has a high quality imbalance (QI) between sample groups, it is likely that technical artifacts are driving the enrichment of these pathways rather than the underlying biology [100].
Troubleshooting Guide: Identifying and Correcting Technical Biases
Problem: Suspect technical bias is influencing pathway enrichment results.
Diagnosis: Follow this systematic workflow to identify potential sources of bias in your data.
Solutions: Based on the diagnostic steps, apply the following corrective actions to your experimental protocol and analysis.
Table 1: Correcting Common Library Preparation Biases [4] [2]
| Bias Category | Root Cause | Corrective Action |
|---|---|---|
| Sample Input / Quality | Degraded RNA or contaminants (phenol, salts). | Re-purify input sample; use fluorometric quantification (Qubit) over absorbance; check 260/230 and 260/280 ratios [4]. |
| Fragmentation & Ligation | Non-random fragmentation; inefficient ligase activity; improper adapter concentration. | Optimize fragmentation time/energy; titrate adapter-to-insert ratio; use fresh ligase buffer [4]. |
| Amplification (PCR) | Too many PCR cycles; polymerase bias for GC-neutral templates. | Use the minimum number of PCR cycles; switch to high-fidelity polymerases (e.g., Kapa HiFi); for GC-rich targets, use additives like betaine [4] [2]. |
| Priming Bias | Uneven reverse transcription with random hexamers. | Use a read-count reweighing scheme in bioinformatics analysis to adjust for the bias [2]. |
Table 2: Mitigating Bias in Pathway Analysis [101] [100]
| Problem | Impact on PEA | Solution |
|---|---|---|
| Quality Imbalance (QI) | Inflates the number of false positive differential genes; enriches for stress-response pathways. | Calculate a QI index for your dataset; remove extreme quality outliers before differential expression analysis [100]. |
| Incorrect Analysis Type | Using an overrepresentation analysis (ORA) on a ranked gene list fails to treasure subtle expression changes. | For ranked gene lists, use a Gene Set Enrichment Analysis (GSEA) approach [101]. |
| Poor Input Gene List | A low-quality or contaminated gene list produces meaningless enrichment results ("garbage in, garbage out"). | Ensure the quality of the input gene list is high before performing enrichment analysis [101]. |
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Bias Mitigation
| Reagent / Tool | Function | Role in Reducing Bias |
|---|---|---|
| High-Fidelity Polymerase (e.g., Kapa HiFi) | PCR amplification of the library. | Reduces amplification bias by amplifying cDNA molecules more uniformly than standard polymerases [2]. |
| Ribonuclease (RNase) Inhibitors | Protects RNA from degradation during extraction and handling. | Prevents RNA degradation, a major source of bias and reduced library complexity [2]. |
| Silica-gel-based Column Kits (e.g., mirVana) | Isolation and purification of RNA, including small RNAs. | Provides higher yields and better quality RNA compared to TRIzol alone, reducing bias in non-coding RNA studies [2]. |
| Fluorometric Quantification Kits (e.g., Qubit) | Accurate measurement of nucleic acid concentration. | Avoids overestimation of usable material by contaminants, a common issue with UV absorbance methods [4]. |
| g:Profiler g:GOSt | A web tool for functional enrichment analysis. | Correctly performs both ORA and rank-based GSEA, allowing users to select the right analysis for their data type [101]. |
Experimental Protocol: Validating Pathway Results
Aim: To confirm that the results of a pathway enrichment analysis are driven by biology and not technical bias.
Procedure:
- Calculate a Quality Imbalance (QI) Index: Using a tool or custom script, calculate the probability of each sample being of low quality based on a machine learning classifier. Then, compute the QI index for your dataset to determine if sample quality is confounded with the experimental groups. A high QI index (e.g., >0.30) indicates a serious risk of technical bias [100].
- Check for Quality Markers: Cross-reference your list of significantly differentially expressed genes with a database of known low-quality marker genes. A significant overlap suggests technical artifacts are influencing your results [100].
- Re-analyze with Outliers Removed: If the previous steps indicate a problem, remove the most severe quality outliers from your dataset and re-run the differential expression and pathway enrichment analysis.
- Compare Results: A robust biological signal should persist after the removal of low-quality samples. If the top-enriched pathways change dramatically or the number of significant genes drops substantially, it strongly suggests the original findings were technically driven [100].
Technical validation is a fundamental requirement in molecular biology research, ensuring that experimental results are accurate, reproducible, and reliable. The noticeable lack of technical standardization remains a significant obstacle in the translation of quantitative PCR (qPCR)-based tests and other molecular assays into clinical practice [102]. This is particularly relevant in the context of RNA-seq library preparation, where the extremely complicated workflow can easily produce biases that damage dataset quality and lead to incorrect interpretation of results [2]. These biases can emerge at multiple stages, including sample preservation, RNA extraction, library construction, and sequencing [2] [3].
The integration of qRT-PCR with orthogonal methods represents a powerful approach to address these challenges. Orthogonal validation, defined as corroborating antibody-based results with data obtained using non-antibody-based methods, provides a robust framework for verifying experimental findings [103]. In the broader context of technical validation, this approach involves using multiple, independent experimental techniques and cross-referencing data to verify results, thereby controlling bias and providing more conclusive evidence of specificity [103]. This comprehensive guide provides troubleshooting resources and validation strategies to help researchers navigate these complex technical challenges.
Understanding qRT-PCR Validation Frameworks
Key Performance Metrics for qRT-PCR Assay Validation
The validation of qRT-PCR assays requires careful assessment of multiple performance characteristics, which should be evaluated based on the context of use and adhere to the "fit-for-purpose" concept [102]. The table below summarizes the essential metrics for proper qRT-PCR validation:
| Performance Characteristic | Definition | Acceptance Criteria Considerations |
|---|---|---|
| Analytical Precision | Closeness of two or more measurements to each other [102] | Established through repeatability and reproducibility testing [102] |
| Analytical Sensitivity | Ability of a test to detect the analyte (usually the minimum detectable concentration or LOD) [102] | Depends on the intended application and required detection limits [102] |
| Analytical Specificity | Ability of a test to distinguish target from nontarget analytes [102] | Must demonstrate detection of target sequence without nonspecific amplification [102] |
| Analytical Trueness/Accuracy | Closeness of a measured value to the true value [102] | Evaluated using reference standards or calibrated controls [102] |
| Diagnostic Sensitivity (TPR) | Proportion of positives that are correctly identified [102] | Depends on clinical or research requirements for disease detection [102] |
| Diagnostic Specificity (TNR) | Proportion of negatives that are correctly identified [102] | Must be determined based on intended use and population [102] |
Experimental Design and Workflow Considerations
The validation process for qRT-PCR assays encompasses multiple critical steps that must be standardized to ensure reliable results. The following workflow diagram illustrates the key stages in qRT-PCR assay validation:
Proper sample acquisition and processing are foundational to successful qRT-PCR validation. Sample preservation method significantly impacts RNA quality, with standard storage involving liquid nitrogen or -80°C freezing, though formalin-fixed paraffin-embedded (FFPE) methods are also used despite introducing challenges like nucleic acid cross-linking and fragmentation [2]. RNA extraction methods must be carefully selected based on the specific application, as standard TRIzol (phenol:chloroform extraction) may cause small RNA loss at low concentrations, while alternative protocols like the mirVana miRNA isolation kit may produce higher yields for certain RNA types [2].
qRT-PCR Troubleshooting Guide
Common Experimental Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Poor Reaction Efficiency [104] | PCR inhibitors, pipetting error, old standard curve [104] | Dilute template to reduce inhibitors; practice proficient pipetting with technical triplicates; prepare standard curve fresh [104] |
| Amplification in No Template Control (NTC) [104] | Template splashing, reagent contamination, primer-dimer formation [104] | Clean work area with 70% ethanol; prepare fresh primer dilution; prevent template splashing; add dissociation curve to detect primer-dimer [104] |
| Inconsistent Biological Replicates [104] | RNA degradation, minimal starting material [104] | Check RNA concentration/quality (260/280 ratio ~1.9-2.0); run RNA on agarose gel; repeat RNA isolation with improved method [104] |
| Ct Values Too Early [104] | Primers not spanning exon-exon junction, genomic DNA contamination, highly expressed transcript, sample evaporation [104] | Design primers spanning exon-exon junctions; DNase treat samples; dilute template; seal tube caps with parafilm [104] |
| No Amplification [105] | Low-abundance target, suboptimal reverse transcription, insufficient cDNA [105] | Increase RNA input; increase cDNA in reaction (max 20% by volume); try different RT kit; consider one-step workflow [105] |
| Unexpected Values [104] | Incorrect instrument protocol, mislabeled samples, wrong dye selection [104] | Check thermal cycling conditions before run; verify correct dyes/volume/wells; use specific user accounts for saved protocols [104] |
Optimization Strategies for Specific Applications
When troubleshooting qRT-PCR experiments, researchers should consider application-specific requirements. For gene expression studies using SYBR Green chemistry, always check melt curves for the number of peaks—when primers are specific, you should see only one peak, while extra peaks could indicate primer-dimers, nonspecific products, or gDNA contamination [105]. For low-abundance targets where sensitivity is problematic (Ct > 32), several approaches can improve detection: increase the amount of RNA input into the reverse transcription reaction, increase the amount of cDNA in the qPCR reaction (up to 20% by volume maximum), try a different reverse transcription kit for higher cDNA yield, or consider a one-step or Cells-to-CT workflow depending on sample type [105].
Orthogonal Methodologies for Technical Validation
Principles of Orthogonal Validation
Orthogonal validation involves cross-referencing experimental results with data obtained using independent, non-antibody-based methods [103]. This approach is similar in principle to using a reference standard to verify a measurement—just as a different, calibrated weight is needed to check if a scale is working correctly, antibody-independent data is required to cross-reference and verify the results of an antibody-driven experiment [103]. The International Working Group on Antibody Validation recommends orthogonal strategies as one of "five conceptual pillars for antibody validation" [103].
Implementing Orthogonal Approaches
The following workflow illustrates how orthogonal validation strategies can be integrated with qRT-PCR to create a comprehensive technical validation framework:
Multiple experimental techniques and public resources can provide orthogonal data for technical validation:
Experimental Techniques:
- RNA-seq: Measures RNA levels through mRNA enrichment, cDNA synthesis, and next-generation sequencing [103]
- Quantitative PCR: Amplifies and quantifies specific DNA sequences using DNA primers [103]
- Mass spectrometry: Identifies and quantifies proteins based on mass-to-charge ratios [103]
- In situ hybridization: Uses labeled nucleic acid probes to detect specific DNA or RNA sequences in tissues or cells [103]
Public Data Resources:
- Human Protein Atlas: Provides RNA normalized expression data across various cell lines and tissues [103]
- Cancer Cell Line Encyclopedia (CCLE): Offers genomic data and analysis for over 1,100 cancer cell lines [103]
- DepMap Portal: Contains datasets identifying cancer vulnerabilities and potential therapeutic targets [103]
- COSMIC (Catalogue Of Somatic Mutations In Cancer): Curated database of somatic mutations across various cancers [103]
- BioGPS: Centralized gene portal aggregating distributed gene annotation resources [103]
Addressing RNA-seq Library Preparation Biases
RNA-seq technologies face various challenges related to bias introduction during library preparation. The table below summarizes major bias sources and recommended improvement strategies:
| Bias Source | Impact on Data Quality | Recommended Improvement Strategies |
|---|---|---|
| Sample Preservation [2] | RNA degradation, cross-linking (especially in FFPE samples) [2] | Use non-cross-linking organic fixatives; for degraded samples, use high input; use random priming instead of oligo-dT [2] |
| RNA Extraction [2] | Small RNA loss, RNA degradation [2] | Use high RNA concentrations; consider alternative protocols (e.g., mirVana kit) [2] |
| mRNA Enrichment [2] | 3'-end capture bias during poly(A) enrichment [2] | Use rRNA depletion instead of poly(A) selection [2] |
| Fragmentation [2] | Reduced complexity from non-random fragmentation [2] | Use chemical treatment (e.g., zinc) rather than RNase III; fragment cDNA instead of RNA [2] |
| Priming Bias [2] | Random hexamer priming bias [2] | Ligate sequencing adapters directly onto RNA fragments; use read count reweighing schemes [2] |
| Adapter Ligation [2] | Substrate preferences of T4 RNA ligases [2] | Use adapters with random nucleotides at ligation extremities [2] |
| PCR Amplification [2] | Preferential amplification of sequences with specific GC content [2] | Use Kapa HiFi rather than Phusion polymerase; reduce amplification cycles; use PCR additives [2] |
Experimental Design Considerations for Minimizing Bias
Thoughtful experimental design is critical for minimizing technical variation in RNA-seq experiments. Technical variation stems from many sources, including differences in RNA quality and quantity, library preparation batch effects, flow cell and lane effects, and adapter bias [71]. To mitigate these issues:
- Replication Strategy: Maintain separate biological replicates rather than pooling samples, as this allows estimation of biological variance and increases power to detect subtle expression changes [71]
- Sample Randomization: Randomize samples during preparation and dilute to the same concentration to minimize batch effects [71]
- Multiplexing Approach: Index and multiplex samples across sequencing lanes to mitigate flow cell and lane effects; when complete multiplexing isn't possible, use a blocking design that includes some samples from each group on each lane [71]
- Library Complexity: Monitor potential PCR duplicates, though true duplicates should be rare; overlapping fragments may occur by chance rather than representing amplification bias [71]
Integrated Validation Workflows
Case Study: Orthogonal Validation in Practice
A representative example of orthogonal validation can be illustrated through the validation of the Nectin-2/CD112 antibody. Researchers first consulted RNA expression data from the Human Protein Atlas to identify cell lines with high (RT4 and MCF7) and low (HDLM-2 and MOLT-4) expression of Nectin-2 RNA [103]. They then performed Western blot analysis using the antibody in these four cell line samples [103]. The results showed elevated protein expression in RT4 and MCF7 and minimal to no expression in HDLM-2 and MOLT-4, confirming correlation between protein expression measured via Western blot and RNA expression data from an orthogonal source [103].
Similarly, for DLL3 (Delta-like ligand 3) antibody validation, researchers used Liquid Chromatography-Mass Spectrometry (LC-MS) data from small cell lung carcinoma samples to identify tissues with high, medium, and low DLL3 peptide counts [103]. Immunohistochemistry analysis using the DLL3 antibody showed protein expression patterns that correlated strongly with the LC-MS peptide counts, with tissues exhibiting minimal, medium, and high abundance staining corresponding to the low, medium, and high peptide counts identified by mass spectrometry [103].
Research Reagent Solutions
The table below outlines essential research reagents and their applications in technical validation workflows:
| Reagent/Kit | Primary Function | Application Context |
|---|---|---|
| mirVana miRNA Isolation Kit [2] | RNA extraction with high yield and quality for noncoding RNAs [2] | Superior to TRIzol for small RNA preservation [2] |
| SuperScript VILO Master Mix [105] | Reverse transcription with high cDNA yield [105] | Ideal for low-abundance targets requiring high sensitivity [105] |
| Kapa HiFi Polymerase [2] | PCR amplification with reduced GC bias [2] | Preferable to Phusion for amplification of GC-rich regions [2] |
| DNase Treatment Reagents [104] | Removal of genomic DNA contamination [104] | Essential step prior to reverse transcription for specific RNA measurement [104] |
| Nuclease-Free Water [104] | Diluent for molecular reactions [104] | Prevents RNA degradation and maintains reaction integrity [104] |
Technical validation through integrated qRT-PCR and orthogonal methods is essential for producing reliable, reproducible research findings. By implementing the troubleshooting guides, addressing RNA-seq biases, and applying orthogonal validation strategies outlined in this technical support center, researchers can significantly enhance the quality and interpretability of their data. The consistent application of these validation principles across experimental workflows represents a critical step toward addressing the reproducibility challenges in molecular biology and translational research.
Troubleshooting Guides
Guide 1: Troubleshooting Low Library Yield
Q: My final RNA-seq library concentration is much lower than expected. What could be causing this and how can I fix it?
Low library yield is a common issue that can stem from multiple points in the preparation workflow. The table below outlines primary causes and corrective actions.
| Primary Cause | Mechanism of Yield Loss | Corrective Action |
|---|---|---|
| Poor Input Quality / Contaminants [4] | Enzyme inhibition (ligases, polymerases) by residual salts, phenol, EDTA, or polysaccharides [4]. | Re-purify input sample; ensure wash buffers are fresh; target high purity (260/230 > 1.8) [4]. |
| Inaccurate Quantification [4] | Under- or over-estimating input concentration leads to suboptimal enzyme stoichiometry [4]. | Use fluorometric methods (Qubit) over UV absorbance; calibrate pipettes; use master mixes [4]. |
| Fragmentation/Tagmentation Inefficiency [4] | Over- or under-fragmentation reduces adapter ligation efficiency or removes library molecules [4]. | Optimize fragmentation time/energy; verify fragmentation profile before proceeding [4]. |
| Suboptimal Adapter Ligation [2] [8] | Poor ligase performance, wrong molar ratio, or reaction conditions reduce adapter incorporation [4]. | Titrate adapter:insert molar ratios; ensure fresh ligase and buffer; maintain optimal temperature [4]. |
| Overly Aggressive Purification [4] | Desired fragments are excluded or lost during bead-based cleanup or size selection [4]. | Use correct bead-to-sample ratio; avoid over-drying beads [4]. |
Guide 2: Addressing PCR Amplification Bias
Q: My data shows high duplicate rates and uneven coverage. How can I minimize amplification bias?
PCR amplification is a major source of bias, where molecules are amplified unevenly, compromising quantitative accuracy [2].
| Primary Cause | Impact on Data | Corrective Action |
|---|---|---|
| Too Many PCR Cycles [4] | High duplicate rates, overamplification artifacts, and flattening of coverage distribution [4]. | Reduce the number of amplification cycles [2] [4]. Use the minimum number needed for library detection. |
| Polymerase Choice [2] | Preferential amplification of fragments with specific GC content, skewing representation [2]. | Use high-fidelity polymerases like Kapa HiFi rather than Phusion [2]. |
| Primer Exhaustion/Mispriming [4] | Dropouts or skew in coverage, particularly for GC-rich or AT-rich regions [4]. | Optimize annealing conditions; for extreme GC content, use additives like TMAC or betaine [2]. |
| Minute Input Quantities [2] | In single-cell or ultra-low input protocols, stochastic effects are magnified [2]. | For single-cell inputs, consider methods like Multiple Displacement Amplification (MDA) [2]. |
Guide 3: Overcoming Library Preparation Bias
Q: What are the main sources of bias during library construction and how can I mitigate them?
Biases introduced during steps like mRNA enrichment and ligation can lead to inaccurate transcript representation [2].
| Source of Bias | Description | Mitigation Strategy |
|---|---|---|
| mRNA Enrichment Bias [2] | Oligo-dT enrichment can introduce 3'-end capture bias, under-representing partially degraded transcripts or those with shorter poly-A tails [2]. | For degraded samples (e.g., FFPE), use rRNA depletion instead of poly-A selection [2] [106]. |
| Adapter Ligation Bias [2] [8] | T4 RNA ligases have sequence-dependent preferences, over-representing fragments that can co-fold with the adaptor [8]. | Use adapters with random nucleotides at the ligation ends [2] or employ single-adaptor circularization methods (CircLigase) [8]. |
| Priming Bias [2] | Random hexamer priming can be non-uniform, leading to uneven coverage across transcripts [2]. | For some applications, directly ligate adapters to RNA fragments, avoiding cDNA synthesis with random primers [2]. |
| Fragmentation Bias [2] | Enzymatic fragmentation (e.g., RNase III) may not be completely random, reducing library complexity [2]. | Use chemical treatment (e.g., zinc) for fragmentation [2] or fragment cDNA post-synthesis [2]. |
Frequently Asked Questions (FAQs)
Q1: How do I choose between oligo(dT) and rRNA depletion for mRNA enrichment?
The choice depends on your sample quality and research goals [106].
- Use oligo(dT) priming when working with high-quality RNA (RIN ≥ 8) and your goal is to profile polyadenylated mRNA. This method is specific but can introduce 3'-bias [2] [106].
- Use rRNA depletion (ribodepletion) when working with degraded RNA (e.g., from FFPE samples), prokaryotic RNA, or when you need to profile both coding and non-coding RNA [2] [106]. This method provides more uniform transcript coverage but can be less efficient at removing ribosomal reads in low-input scenarios [106].
Q2: What is the minimum RNA input required for a successful RNA-seq library, and what options exist for low-input samples?
Standard protocols may require 100-500 ng of total RNA, but many specialized kits are designed for low-input and single-cell applications [59] [106].
- Ultra-low input (10 pg - 10 ng): Kits like the SMART-Seq v4 or SMARTer Ultra Low use template-switching technology to generate full-length cDNA from intact cells or high-quality RNA [106].
- Degraded/low-quality RNA (200 pg - 10 ng): For samples with low RIN (e.g., from FFPE), the SMARTer Stranded or SMARTer Universal Low Input kits are recommended. These use random priming and require prior rRNA depletion [106].
Q3: How does the choice of library preparation kit impact gene expression results?
Different kits can introduce protocol-specific biases, but studies show good correlation for overall gene expression. A 2022 comparative analysis of Illumina TruSeq, Swift, and Swift Rapid kits found that normalized gene expression measurements were highly correlated (Pearson correlation > 0.97) across methods [59]. The main differences often lie in the detection of the lowest abundance transcripts, workflow time, and cost [59] [107]. It is critical to use the same kit and protocol for all samples within a single study.
Q4: What are some cost-effective strategies for high-throughput RNA-seq library preparation?
For labs processing many samples, consider:
- Tn5 Transposase-based "Tagmentation": Protocols using homemade or commercial Tn5 transposase combine fragmentation and adapter ligation into a single step, significantly reducing time and cost (e.g., under £10 per sample) [107].
- Automation: Using liquid handling robots for library preparation increases throughput, improves reproducibility, and minimizes human error [59].
- Bulk Reagents: Purchasing enzymes and buffers in bulk rather than in individual kit formats can lead to substantial savings [107].
Experimental Protocols for Bias Evaluation
Protocol 1: Comparative Analysis of Library Prep Kits
Objective: To systematically evaluate the performance and bias of different RNA-seq library preparation kits using a common reference RNA sample.
Materials:
- Universal Human Reference RNA (UHRR) [59]
- Library preparation kits for comparison (e.g., Illumina TruSeq, Swift RNA, Swift Rapid RNA) [59]
- Equipment for quality control (e.g., Bioanalyzer, Qubit fluorometer) [4]
- Illumina sequencer
Methodology:
- Sample Allocation: Aliquot the same UHRR sample to different input amounts recommended by each kit's protocol (e.g., 10 ng, 50 ng, 100 ng, 500 ng) [59].
- Library Preparation: Prepare libraries in replicate (n=5) for each kit and input amount combination, strictly following manufacturers' protocols [59].
- Quality Control: Assess final library quality using an Bioanalyzer for fragment size distribution and a fluorometer for accurate quantification [4] [59].
- Sequencing: Sequence all libraries to a standardized depth (e.g., 20 million reads per library) on the same sequencing platform [59].
- Bioinformatic Analysis:
- Alignment: Map reads to the reference genome using a robust aligner (e.g., STAR).
- Correlation Analysis: Calculate the correlation of normalized gene counts (e.g., using regularized log transform) between kits. High agreement is indicated by Pearson correlation >0.97 [59].
- Differential Expression: Identify genes that are differentially expressed between kits, which indicates protocol-specific bias [59].
- Coverage Uniformity: Assess 5'/3' coverage bias using tools like Picard Tools [59].
Protocol 2: Evaluating Ligation Bias with a Defined RNA Pool
Objective: To quantify sequence-specific bias introduced during the adapter ligation step of library construction [8].
Materials:
- Defined pool of synthetic RNA oligonucleotides (with a degenerate region) [8]
- Components for library preparation: T4 RNA Ligase 2, CircLigase, Mth K97A ligase, etc. [8]
- PCR purification kits
- Sequencing platform (e.g., Ion Torrent PGM)
Methodology:
- Library Construction: Divide the defined RNA pool and prepare sequencing libraries using different ligation protocols or enzymes (e.g., standard duplex adaptor vs. CircLigase single adaptor protocol) [8].
- Sequencing: Sequence the final libraries.
- Bias Quantification:
- Theoretical Distribution: Calculate the expected read count for each unique sequence in the degenerate pool, assuming perfectly uniform representation.
- Observed Distribution: Count the actual reads for each sequence.
- Over-representation Analysis: Identify sequences that are significantly over- or under-represented compared to the theoretical expectation. The CircLig protocol has been shown to reduce over-representation compared to standard protocols [8].
- Sequence/Structure Analysis: Correlate over-representation with sequence features (e.g., GC content) or predicted secondary structure and potential for co-folding with the adapter [8].
Experimental Workflow and Bias Pathways
Decision Framework for RNA-seq Library Prep
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Kit | Primary Function | Key Considerations for Bias Reduction |
|---|---|---|
| SMART-Seq v4 Ultra Low Input Kit [106] | Full-length cDNA synthesis from ultra-low input (1-1,000 cells) or high-quality RNA (RIN ≥8) using template-switching and oligo-dT priming. | Improves coverage of GC-rich transcripts. Ideal for minimizing bias when cell numbers are limited but RNA is intact [106]. |
| SMARTer Stranded RNA-Seq Kit [106] | Preparation of strand-specific libraries from degraded or low-quality RNA (e.g., FFPE). Uses random priming. | Requires prior rRNA depletion. Maintains strand information with >99% accuracy, crucial for accurate assignment of reads in overlapping genes [106]. |
| RiboGone Depletion Kit [106] | Depletes ribosomal RNA from mammalian total RNA samples (10-100 ng). | Essential for library prep from degraded samples or when using random-primed kits to prevent >90% of reads from mapping to rRNA [106]. |
| Kapa HiFi Polymerase [2] | High-fidelity PCR amplification during library enrichment. | Reduces preferential amplification biases associated with GC-rich or AT-rich regions compared to other polymerases like Phusion [2]. |
| User-Prepared Tn5 Transposase [107] | Simultaneously fragments cDNA and ligates adapters ("tagmentation"). | A low-cost, high-throughput alternative to kit-based fragmentation/ligation. Streamlines workflow and reduces hands-on time [107]. |
| CircLigase [8] | Single-stranded DNA ligase used in circularization-based library protocols. | Significantly reduces ligation bias compared to standard duplex adaptor protocols using T4 RNA ligase [8]. |
Conclusion
RNA-seq library preparation biases are not merely technical nuisances but fundamental considerations that directly impact biological interpretation and translational potential. A strategic approach combining informed kit selection based on experimental needs, rigorous quality control, and appropriate validation is paramount for generating reliable data. Future directions should focus on developing更低偏倚的ligation methods, improved normalization strategies using spike-ins, and standardized benchmarking protocols that enable cross-study comparisons. As RNA-seq applications expand into clinical diagnostics and drug development, acknowledging and mitigating these biases becomes increasingly critical for deriving biologically meaningful insights and advancing precision medicine initiatives.
References
- 1. Almost all steps of NGS library preparation protocols ... [https://www.rna-seqblog.com/almost-all-steps-of-ngs-library-preparation-protocols-introduce-bias-especially-in-the-case-of-rna-seq-2/]
- 2. Bias in RNA-seq Library Preparation: Current Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8079181/]
- 3. Bias in RNA-seq Library Preparation [https://www.rna-seqblog.com/bias-in-rna-seq-library-preparation/]
- 4. How to Troubleshoot Sequencing Preparation Errors (NGS ... [https://www.cd-genomics.com/resource-sequencing-preparation-troubleshooting.html]
- 5. 5 Best Practices for NGS Library Prep Success [https://dispendix.com/blog/5-best-practices-for-ngs-library-prep-success]
- 6. Identification and remediation of biases in the activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3241666/]
- 7. Bias in Ligation-Based Small RNA Sequencing Library ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0126049]
- 8. Evaluating bias-reducing protocols for RNA sequencing ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-569]
- 9. Addressing Bias in Small RNA Library Preparation for ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2015.00352/full]
- 10. Troubleshooting Guide for Ligases [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/troubleshooting-guide-for-ligases?srsltid=AfmBOorzVzYrXzfsHGyQg8RZXGyesCL_Y6MO3kj_ncHJBJkebKpsaNO6]
- 11. Optimize Your DNA Ligation with 7 Must-Have Tips [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/optimize-dna-ligase.html]
- 12. Evaluating Quality of Input RNA for NGS Library Preparation [https://www.zymoresearch.com/pages/technote-evaluating-quality-of-input-rna-for-ngs-library-preparation?srsltid=AfmBOopenolIt0elusmUIG-exytHaRJFS2IK6f75o6dXvobQSPzUu5iI]
- 13. A head-to-head comparison of ribodepletion and polyA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8495925/]
- 14. Evaluation of two main RNA-seq approaches for gene ... [https://www.nature.com/articles/s41598-018-23226-4]
- 15. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-419]
- 16. Ribosomal RNA Depletion [https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/rna-extraction/rna-applications/ribosomal-rna-depletion.html]
- 17. Choosing Poly(A) Selection or rRNA Depletion for RNA-seq [https://www.revvity.com/blog/polya-selection-vs-rrna-depletion-guide-picking-right-upstream-method]
- 18. Comparison of rRNA depletion methods for efficient ... [https://www.nature.com/articles/s41598-022-09710-y]
- 19. NEBNext® RNA Library Prep Kit Troubleshooting Guide ... [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/nebnext-ultratm-directional-rna-kit-troubleshooting-guide?srsltid=AfmBOopnS7Hnc-GEvG4FCcMrjSUyiRjLyv1LzAigXxi72MbsjMfsBOqD]
- 20. RNA-Seq Frequently Asked Questions [https://web.genewiz.com/faqs/rna-seq]
- 21. Paired rRNA-depleted and polyA-selected RNA ... [https://www.nature.com/articles/s41597-020-00719-4]
- 22. SARS-CoV-2 sequencing artifacts associated with targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530606/]
- 23. On the causes, consequences, and avoidance of PCR ... [https://pubmed.ncbi.nlm.nih.gov/37062860/]
- 24. Identification and removal of sequencing artifacts produced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6097653/]
- 25. How to deal with PCR artifacts in RNASeq data? [https://www.biostars.org/p/276250/]
- 26. A computational method for estimating the PCR duplication ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1471-9]
- 27. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 28. New England Biolabs® launches NEBNext® Low-bias ... [https://www.neb.com/en-us/about-neb/news-and-press-releases/new-england-biolabs-launches-nebnext-low-bias-small-rna-library-prep-kit-presenting-a-new-method-for-capturing-the-true-diversity-of-rna-samples?srsltid=AfmBOop2sNxUqoJKcNKlhqbjzCx3cGav6DeGhXw5dM-le-WpLWiHtAuV]
- 29. RNA-seq FAQs [https://www.takarabio.com/learning-centers/next-generation-sequencing/rna-seq/rna-seq-faqs?srsltid=AfmBOoqQRAZ4U0L5P5RUvlWu8T1mb8uVo-h2OH2kRuTV1zpefMoS7p6j]
- 30. RNA-SeQC: RNA-seq metrics for quality control and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3356847/]
- 31. Reverse transcription troubleshooting guide [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/rt-education/reverse-transcription-troubleshooting.html]
- 32. Stranded vs Non-Stranded RNA-Seq [https://blog.genewiz.com/stranded-vs-non-stranded-rna-seq]
- 33. Comparison of Stranded and Non-Stranded RNA ... [https://rna.cd-genomics.com/resource/stranded-and-non-stranded-rna-sequencing.html]
- 34. Comparison of stranded and non-stranded RNA-seq ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-1876-7]
- 35. Commentary: a review of technical considerations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522984/]
- 36. Bioinformatics for RNA‐Seq Data Analysis [https://www.intechopen.com/chapters/50574]
- 37. The impact of PCR duplication on RNAseq data generated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12117910/]
- 38. Challenges in Single-Cell RNA Seq Data Analysis & ... [https://www.elucidata.io/blog/challenges-and-solutions-in-single-cell-rna-seq-data-analysis]
- 39. Optimizing RNA-Seq Strategies for Transcriptomic Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6288342/]
- 40. Improving the sensitivity of ultra-low input mRNA-seq [https://www.takarabio.com/learning-centers/next-generation-sequencing/rna-seq/technotes/full-length-single-cell-library-method-comparison?srsltid=AfmBOoqujYDs-XLrb2_ATyxG3l-Xa8PXGGcok0pZym1sB3vV-LHSxA9W]
- 41. NEBNext® RNA Library Prep Kit Troubleshooting Guide ... [https://www.neb.com/en/tools-and-resources/troubleshooting-guides/nebnext-ultratm-directional-rna-kit-troubleshooting-guide?srsltid=AfmBOorkBkqf3LCZO5JwE7uScZKrZxAvppBhgxMvSqxJjFlMyID90gZd]
- 42. Guide for library design and bias correction for large-scale ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3017-9]
- 43. RNA-seq FAQs [https://www.takarabio.com/learning-centers/next-generation-sequencing/rna-seq/rna-seq-faqs?srsltid=AfmBOoqLZgnkH2jnQm32il6I-nMWhrG0J65ADFyNolLPz064EPNnuXec]
- 44. NEBNext® RNA Library Prep Kit Troubleshooting Guide ... [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/nebnext-ultratm-directional-rna-kit-troubleshooting-guide?srsltid=AfmBOoqUht9QrPYXukW24TsHUajjmZylqCqQ1Usg0c7o2BwmzrvCDCFM]
- 45. Swift Biosciences Partners with MGI for High Throughput ... [https://en.mgi-tech.com/news/127/]
- 46. Troubleshooting Guide for use with the NEBNext® RNA ... [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/troubleshooting-guide-for-use-with-the-nebnext-rna-depletion-core-reagent-set-and-nebnext-custom-rna-depletion-tool?srsltid=AfmBOoprVnCTWlFFlqlw9YW6kbjGdUL3Kl-OTi2xH0HWflLC65p6ewxS]
- 47. Addressing Bias in Small RNA Library Preparation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4686641/]
- 48. Systematic comparison of quantity and quality of RNA recovered with commercial FFPE tissue extraction kits | Journal of Translational Medicine [https://link.springer.com/article/10.1186/s12967-024-05890-5]
- 49. Overcoming challenges in FFPE DNA library prep [https://www.neb.com/en-us/nebinspired-blog/overcoming-challenges-in-ffpe-dna-library-prep?srsltid=AfmBOopWekcuipwXTYrjclWg3VVbbNsTJr20xKKNkMHsNiIzM5wNzf2A]
- 50. Enhanced quality metrics for assessing RNA derived from archival formalin-fixed paraffin-embedded samples - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7029400/]
- 51. Quality control recommendations for RNASeq using FFPE samples based on pre-sequencing lab metrics and post-sequencing bioinformatics metrics | BMC Medical Genomics | Full Text [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-022-01355-0]
- 52. RNA-seq: impact of RNA degradation on transcript quantification [https://pmc.ncbi.nlm.nih.gov/articles/PMC4071332/]
- 53. Impact of RNA degradation on next-generation sequencing ... [https://www.sciencedirect.com/science/article/pii/S0888754322001744]
- 54. Whole Transcriptome vs 3' mRNA-Seq: How to Choose ... [https://www.lexogen.com/blog/choosing-between-whole-transcriptome-total-rna-seq-and-3-mrna-seq/]
- 55. A comparison between whole transcript and 3' RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-5393-3]
- 56. When do you recommend 3'-Tag RNA-seq? [https://dnatech.ucdavis.edu/faqs/when-do-you-recommend-3-tag-rna-seq]
- 57. Full-Length BRB-seq vs Standard Full-Length mRNA-seq [https://alitheagenomics.com/blog/what-is-full-length-brb-seq-and-how-well-does-it-perform]
- 58. What is 3' mRNA sequencing? [https://alitheagenomics.com/blog/what-is-3-mrna-sequencing]
- 59. Systematic comparative analysis of strand-specific RNA ... [https://www.nature.com/articles/s41598-021-04583-z]
- 60. end mRNA library from unpurified bulk RNA in a single tube [https://www.nature.com/articles/s12276-024-01164-8]
- 61. Benchmarking full-length transcript single cell mRNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-09014-5]
- 62. Overview of Published 3' mRNA-seq Workflows [https://alitheagenomics.com/blog/overview-of-published-3-mrna-seq-workflows-from-early-multiplexing-and-beyond]
- 63. What is the RNA Integrity Number (RIN)? [https://www.cd-genomics.com/longseq/resource-rna-integrity-number.html]
- 64. The Importance of Quality Control in RNA-Seq Analysis [https://ngscloud.com/the-importance-of-quality-control-in-rna-seq-analysis/]
- 65. RNA integrity number [https://en.wikipedia.org/wiki/RNA_integrity_number]
- 66. RNA Integrity Number - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/rna-integrity-number]
- 67. 3' Bias in RNASEQ data [https://www.biostars.org/p/191442/]
- 68. Integration of Bulk RNA-seq Pipeline Metrics for Assessing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236924/]
- 69. 5 tips for successful single-cell RNA-seq [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/5-tips-to-make-your-single-cell-rna-seq-experiments-a-success?srsltid=AfmBOooBlKki1ZYkrUczPS47o4hHJMnAj_b_AcVArWLv7yLS3OXcCT9y]
- 70. Development of an End-to-End Total RNA Sequencing ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157825001436]
- 71. RNA-seq Data: Challenges in and Recommendations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4230301/]
- 72. EFFICIENT RIBOSOMAL RNA DEPLETION FROM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123297/]
- 73. Effective ribosomal RNA depletion for single-cell total ... [https://peerj.com/articles/10717/]
- 74. Improving an rRNA depletion protocol with statistical ... [https://www.sciencedirect.com/science/article/pii/S2472630322051767]
- 75. Assessing and mitigating batch effects in large-scale omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11447944/]
- 76. RNA-Seq Experimental Design Guide for Drug Discovery [https://www.lexogen.com/blog/planning-for-success-a-strategic-design-guide-for-rna-seq-experiments-in-drug-discovery/]
- 77. Correcting batch effects in large-scale multiomics studies ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03047-z]
- 78. Highly effective batch effect correction method for RNA-seq ... [https://www.sciencedirect.com/science/article/pii/S200103702400432X]
- 79. An order-preserving batch-effect correction method based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12207412/]
- 80. Comparative analysis of library preparation approaches for ... [https://www.nature.com/articles/s41598-025-12992-7]
- 81. Improving RNA-Seq Library Preparation for Accuracy [https://cellcarta.com/science-hub/rna-seq-library-preparation-watchmaker-workflow/]
- 82. Deciphering RNA-seq Library Preparation: From Principles ... [https://www.cd-genomics.com/resouce-rna-seq-library-preparation-principles-protocol.html]
- 83. How to Improve Your RNA-Seq Data with UMIs and ERCC ... [https://blog.genewiz.com/how-to-improve-your-rna-seq-data-with-umis-and-ercc-rna]
- 84. SIRVs (Spike-in RNA Variant Control Mixes) [https://www.lexogen.com/sirvs/]
- 85. SIRVs: RNA-seq controls from @Lexogen [http://enseqlopedia.com/2016/10/sirvs-rna-seq-controls-from-lexogen/]
- 86. SIRV-Sets | Spike-In RNA Variant Control Mixes [https://www.lexogen.com/sirvs/sirv-sets/]
- 87. Analyzing RNA Seq splice variants using spike-ins? [https://www.biostars.org/p/174688/]
- 88. Problems using ERCC spike-ins for normalization in DESeq2 [https://www.biostars.org/p/9573278/]
- 89. Assessing the reliability of spike-in normalization for analyses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5668938/]
- 90. Essential RNA-seq Metrics: Part 1 [https://www.rna-seqblog.com/essential-rna-seq-metrics-part-1/]
- 91. Comprehensive comparative analysis of RNA sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3821180/]
- 92. Why total RNA-seq usually yields low mapping rate? [https://bioinformatics.stackexchange.com/questions/2897/why-total-rna-seq-usually-yields-low-mapping-rate]
- 93. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 94. RNA Sequencing Quality Control [https://rna.cd-genomics.com/resource/rna-sequencing-quality-control.html]
- 95. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 96. Differentially expressed genes from RNA-Seq and functional enrichment results are affected by the choice of single-end versus paired-end reads and stranded versus non-stranded protocols | BMC Genomics | Full Text [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3797-0]
- 97. Selecting between-sample RNA-Seq normalization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6171491/]
- 98. RNA-Seq Normalization: Methods and Stages [https://bigomics.ch/blog/why-how-normalize-rna-seq-data/]
- 99. Technical bias and the reproducibility crisis [https://pmc.ncbi.nlm.nih.gov/articles/PMC7857423/]
- 100. Overlooked poor-quality patient samples in sequencing data ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03331-6]
- 101. Nine quick tips for pathway enrichment analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9371296/]
- 102. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 103. Antibody Validation Essentials: Orthogonal Strategy - CST Blog [https://blog.cellsignal.com/hallmarks-of-validation-orthogonal-strategy]
- 104. qPCR Troubleshooting Guide [https://azurebiosystems.com/qpcr-resources/troubleshooting/]
- 105. Real-Time PCR Basics Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/real-time-digital-PCR-applications-support-center/real-time-pcr-basics-support/real-time-pcr-basics-support-troubleshooting.html]
- 106. RNA-seq FAQs [https://www.takarabio.com/learning-centers/next-generation-sequencing/rna-seq/rna-seq-faqs?srsltid=AfmBOorCHu5r32K7OCVuHpsscpNbQN6gELzMUZ2K2zolMtDmAFHX2LIF]
- 107. Low-cost and High-throughput RNA-seq Library ... [https://en.bio-protocol.org/en/bpdetail?id=3799&type=0]