A Comprehensive Guide to Single-Cell RNA Sequencing Protocols: From Experimental Design to Data Validation
This article provides a complete roadmap for researchers and drug development professionals designing single-cell RNA sequencing (scRNA-seq) experiments.
A Comprehensive Guide to Single-Cell RNA Sequencing Protocols: From Experimental Design to Data Validation
Abstract
This article provides a complete roadmap for researchers and drug development professionals designing single-cell RNA sequencing (scRNA-seq) experiments. It covers foundational principles, from understanding cellular heterogeneity and the critical importance of Unique Molecular Identifiers (UMIs) to selecting the appropriate isolation method for your sample type. The guide details step-by-step methodological workflows for both high- and low-throughput platforms, offers solutions for common pitfalls in tissue dissociation and batch effects, and outlines essential techniques for validating scRNA-seq findings through spatial transcriptomics, protein-level assays, and functional studies. By integrating established best practices with the latest advancements, this resource aims to empower scientists to generate robust, reproducible, and biologically insightful single-cell data.
Understanding Single-Cell RNA Sequencing: Core Principles and Experimental Foundations
Single-cell RNA sequencing (scRNA-seq) has emerged as a transformative technology that enables the profiling of gene expression at the ultimate level of biological resolution: the individual cell. Since its conceptual and technical breakthrough in 2009, scRNA-seq has fundamentally altered our ability to dissect cellular heterogeneity, revealing complex cellular landscapes that were previously obscured by bulk sequencing approaches [1] [2]. While traditional bulk RNA sequencing measures the average expression of genes across thousands to millions of cells, scRNA-seq captures the unique transcriptional profile of each individual cell, allowing researchers to identify rare cell populations, characterize novel cell types, and understand regulatory relationships between genes in a way that was never before possible [3].
The fundamental limitation of bulk sequencing lies in its inherent averaging effect. When tissue samples are processed as bulk populations, the resulting transcriptome represents a composite signal from all constituent cells, effectively masking the true biological diversity present within the sample [3]. This averaging can obscure critical biological phenomena, including rare but functionally important cell populations, continuous transitional states in developmental processes, and stochastic expression variations that may drive cell fate decisions [2]. scRNA-seq overcomes these limitations by separately profiling the transcriptomes of individual cells, enabling the direct observation and quantification of cellular heterogeneity within seemingly homogeneous populations [1].
Key Technological Advances Enabling scRNA-seq
Evolution of scRNA-seq Platforms and Methods
The rapid advancement of scRNA-seq has been driven by continuous improvements in both wet-lab methodologies and computational analytics. The core procedure involves isolating single cells, converting their RNA into complementary DNA (cDNA), amplifying the cDNA, and preparing sequencing libraries [1]. Early methods utilized limiting dilution, fluorescence-activated cell sorting (FACS), or micromanipulation for cell isolation, but these approaches were limited in throughput and efficiency [3]. The field has since evolved toward high-throughput platforms capable of processing tens of thousands of cells in a single experiment, with dramatic reductions in cost and increased automation [1].
Two primary amplification strategies have been developed for scRNA-seq: polymerase chain reaction (PCR)-based methods (e.g., SMART-seq2, Fluidigm C1, Drop-seq, 10x Genomics) and in vitro transcription (IVT)-based methods (e.g., CEL-seq, MARS-Seq) [1]. PCR amplification represents a non-linear amplification process that can generate sufficient material for sequencing from the minute quantities of RNA present in a single cell. The introduction of unique molecular identifiers (UMIs) has been particularly valuable for addressing amplification biases, as these random barcodes label individual mRNA molecules during reverse transcription, enabling accurate quantification by effectively distinguishing between biological duplicates and PCR duplicates [1] [2].
Table 1: Comparison of Major scRNA-seq Technological Approaches
| Method Type | Examples | Amplification Strategy | Throughput | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Plate-based | SMART-seq2, Fluidigm C1 | PCR-based (mostly) | Low to medium (96-800 cells) | Full-length transcript coverage; good sensitivity | Lower throughput; higher cost per cell |
| Droplet-based | 10x Genomics, inDrop, Drop-seq | PCR-based | High (thousands to millions of cells) | High throughput; cost-effective | 3' or 5' end counting only; shorter reads |
| Nanowell-based | Seq-Well, Microwell-seq | PCR-based | High (thousands to tens of thousands) | Portable; cost-effective | Lower mRNA capture efficiency |
| IVT-based | CEL-seq, MARS-seq | Linear amplification (IVT) | Medium to high | Better quantitative accuracy | 3' bias; more complex protocol |
Single-Cell Isolation Techniques
The initial step of isolating viable single cells is critical for successful scRNA-seq experiments. The choice of isolation method depends on the tissue type, cell properties, and research objectives [1]. The most common techniques include:
- Limiting Dilution: A traditional method where cell suspensions are diluted to statistically distribute individual cells into multi-well plates [3]. While simple and requiring no specialized equipment, this approach is inefficient with approximately one-third of wells typically containing cells when diluting to 0.5 cells per aliquot [3].
- Fluorescence-Activated Cell Sorting (FACS): Currently the most widely used method, FACS enables the isolation of highly purified single cells based on fluorescently labeled antibodies targeting specific surface markers [3]. This method offers high purity and the ability to select specific cell populations but requires large starting cell numbers and may induce cellular stress [3].
- Microfluidic Technologies: These systems, including the commercial Fluidigm C1 platform, utilize microscale chambers for automated cell processing with minimal reagent volumes [3]. Microfluidics offers improved precision and reduced contamination risk but may be limited by cell size restrictions and lower throughput compared to newer methods [3].
- Droplet-Based Microfluidics: High-throughput platforms like the 10x Genomics Chromium system encapsulate individual cells in nanoliter-sized droplets containing barcoded beads [1]. This approach enables massive parallel processing of thousands to millions of cells at reduced cost, making it ideal for comprehensive atlas projects and detecting rare cell types [1] [2].
A critical consideration in single-cell isolation is the potential for inducing "artificial transcriptional stress responses" during tissue dissociation [1]. Studies have demonstrated that dissociation protocols, particularly those employing proteases at 37°C, can artificially alter transcriptional patterns and lead to inaccurate cell type identification [1]. To minimize these technical artifacts, dissociation at lower temperatures (e.g., 4°C) or utilization of single-nucleus RNA sequencing (snRNA-seq) has been recommended, especially for sensitive tissues like brain [1].
Computational Analysis of scRNA-seq Data
Standard Bioinformatics Pipeline
The analysis of scRNA-seq data presents unique computational challenges due to its high dimensionality, technical noise, and sparsity [4] [3]. A standard analytical workflow encompasses multiple processing steps:
Quality Control and Filtering: The initial critical step involves removing low-quality cells that could compromise downstream analyses. Quality control typically examines three key metrics: the number of counts per barcode (count depth), the number of genes per barcode, and the fraction of mitochondrial counts per barcode [4]. Cells with low counts/genes may represent broken cells or empty droplets, while those with unexpectedly high counts may be doublets (multiple cells). High mitochondrial fractions often indicate compromised cell viability [4]. These metrics should be considered jointly rather than in isolation to avoid unintentionally filtering out biologically relevant cell populations [4].
Normalization and Batch Effect Correction: Normalization addresses differences in sequencing depth between cells, typically by scaling counts to counts per million (CPM) or similar metrics [5]. As datasets often combine cells from multiple experiments, batch effect correction methods are essential to remove technical variations unrelated to biological signals, enabling valid comparisons across datasets [4].
Feature Selection and Dimensionality Reduction: scRNA-seq data typically measure expression of thousands of genes across thousands of cells, creating a high-dimensional space. Dimensionality reduction techniques like principal component analysis (PCA) identify the most informative sources of variation [5]. Further non-linear methods such as t-distributed stochastic neighbor embedding (t-SNE) and Uniform Manifold Approximation and Projection (UMAP) project cells into 2D or 3D spaces for visualization [4] [6].
Clustering and Cell Type Identification: Unsupervised clustering algorithms (e.g., Leiden, Louvain) group cells based on transcriptional similarity, revealing distinct cell populations [5]. Cluster identity is then inferred through differential expression analysis, comparing each cluster against others to find marker genes that can be matched to known cell types using reference databases [4].
Trajectory Inference: For developing systems or continuous biological processes, trajectory inference methods (pseudotemporal ordering) reconstruct the dynamic transitions cells undergo, such as differentiation pathways, by ordering cells along a continuum based on transcriptional similarity [2].
Comparing and Integrating scRNA-seq Datasets
As the number of publicly available scRNA-seq datasets grows, methods for comparing and integrating them have become increasingly important. Two main approaches exist for cross-dataset analysis [7]:
Label-Centric Comparison: This approach focuses on identifying equivalent cell types/states across datasets by comparing individual cells or groups of cells. Methods like scmap project cells from a new experiment onto an annotated reference dataset, similar to BLAST for sequence data, enabling rapid annotation and identification of novel cell states [7].
Cross-Dataset Normalization: These methods computationally remove experiment-specific technical and biological effects so that data from multiple experiments can be combined and analyzed jointly. This approach facilitates the identification of rare cell types that may be too sparsely sampled in individual datasets to be reliably detected [7].
Newer tools like scCompare utilize correlation-based mapping to transfer phenotypic identities from a reference dataset to a query dataset, with statistical thresholds to identify cells distinct from known phenotypes, enabling novel cell type discovery [5].
Applications: Revealing Biological Heterogeneity
Characterizing Cellular Diversity in Tissues and Organs
One of the most significant applications of scRNA-seq has been the comprehensive cataloging of cell types across tissues and organisms through atlas projects like the Human Cell Atlas, Tabula Muris, and Tabula Sapiens [1] [8]. These initiatives aim to create reference maps of all human cells, providing foundational resources for understanding healthy physiology and disease mechanisms. For example, the Tabula Sapiens has collected scRNA-seq data from 24 tissues across multiple donors, enabling cross-tissue comparisons and the identification of tissue-specific and shared cell populations [5].
In immunology, scRNA-seq has revealed unprecedented complexity within immune cell populations, identifying novel subsets of T cells with distinct functional specializations and activation states [6] [2]. Similarly, in neuroscience, scRNA-seq has transformed our understanding of brain complexity by characterizing dozens of neuronal and glial cell types that were previously indistinguishable [1] [3].
Unraveling Disease Mechanisms
In cancer research, scRNA-seq has demonstrated exceptional utility for characterizing tumor heterogeneity, the tumor microenvironment, and cellular mechanisms underlying treatment resistance [2] [3]. By profiling individual cells within tumors, researchers have identified rare subpopulations of therapy-resistant cells that persist after treatment and eventually drive relapse [3]. scRNA-seq of circulating tumor cells (CTCs) has also provided insights into metastasis mechanisms through minimally invasive liquid biopsies [3].
Protocols have been developed for applying scRNA-seq to challenging clinical samples, including bladder Ewing sarcoma, demonstrating the feasibility of generating high-quality data from difficult tumor tissues [9] [10]. Spatial transcriptomics technologies now complement scRNA-seq by preserving the spatial context of gene expression, bridging the gap between cellular heterogeneity and tissue architecture [9].
Tracing Developmental Trajectories
scRNA-seq is ideally suited for studying dynamic biological processes such as embryonic development, tissue regeneration, and cell differentiation [2]. By capturing cells at different stages along a developmental continuum, researchers can reconstruct lineage relationships and identify key transcriptional regulators driving fate decisions [2]. For example, studies of cardiomyocyte differentiation from human induced pluripotent stem cells (hiPSCs) using scRNA-seq have revealed how different protocols generate distinct cellular outcomes, enabling optimization of differentiation methods for regenerative medicine applications [5].
Table 2: Key Research Reagent Solutions for scRNA-seq Experiments
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Cell Isolation Reagents | Enzymatic dissociation cocktails (collagenase, trypsin), FACS antibodies, Dead cell removal kits | Tissue dissociation and viable cell isolation | Optimization needed for each tissue type; minimize stress responses |
| Library Preparation Kits | 10x Genomics Chromium, SMART-seq kits, Fluidigm C1 reagents | Single-cell capture, barcoding, and library construction | Throughput, cost, and transcript coverage vary by platform |
| Reverse Transcriptase Enzymes | Moloney Murine Leukemia Virus (MMLV) RT with template-switching activity | cDNA synthesis from single-cell RNA | High efficiency needed due to low mRNA input |
| Amplification Reagents | PCR master mixes, In vitro transcription kits | cDNA amplification for sequencing library construction | Choice affects 3' bias and quantitative accuracy |
| Unique Molecular Identifiers (UMIs) | Barcoded oligonucleotides | Molecular counting and elimination of PCR duplicates | Essential for accurate quantification |
| Quality Control Assays | Bioanalyzer tapes, Fluorescent cell viability dyes, Ribonuclease inhibitors | Assessment of RNA quality, cell viability, and sample integrity | Critical for preventing experimental failure |
The expansion of scRNA-seq has been accompanied by the development of specialized databases and resources that facilitate data sharing, exploration, and reuse:
- Single Cell Portal (Broad Institute): A specialized platform for sharing and exploring scRNA-seq datasets with built-in visualization tools [8].
- CZ Cell x Gene Discover: Hosts over 500 scRNA-seq datasets with intuitive exploration capabilities through an open-source data exploration tool [8].
- Single Cell Expression Atlas (EMBL-EBI): Provides consistently processed and annotated scRNA-seq datasets across multiple organisms [8].
- PanglaoDB: A curated database of scRNA-seq datasets with annotated marker genes and automated cell type annotations [8].
- scRNAseq Package (Bioconductor): Provides programmatic access to dozens of curated scRNA-seq datasets in standardized formats for easy integration with analytical workflows [8].
These resources have become invaluable for researchers seeking to contextualize their findings within larger biological frameworks, generate hypotheses, and conduct meta-analyses across multiple studies [8].
Single-cell RNA sequencing has fundamentally transformed our approach to studying complex biological systems by providing unprecedented resolution into cellular heterogeneity. As the technologies continue to mature with improvements in throughput, sensitivity, and spatial context preservation, and as computational methods become more sophisticated and standardized, scRNA-seq is poised to become an increasingly integral component of both basic biological research and clinical applications. The ability to deconvolute tissue complexity, identify rare cell populations, track developmental trajectories, and understand disease mechanisms at single-cell resolution will continue to drive discoveries across fields from immunology to cancer biology to regenerative medicine, firmly establishing scRNA-seq as an essential tool for modern biological research.
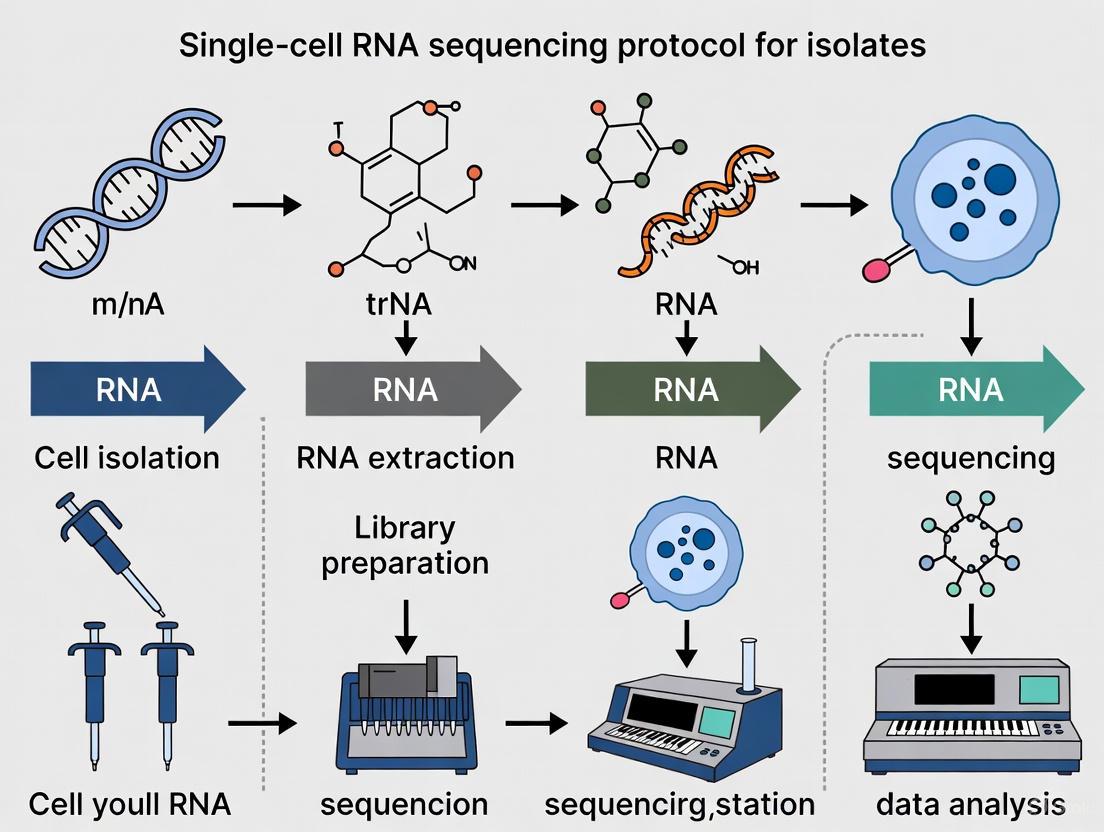
Single-cell RNA sequencing (scRNA-seq) has revolutionized transcriptomic research by enabling the analysis of gene expression patterns at the level of the individual cell. This powerful genomic approach provides an unprecedented view of cellular heterogeneity and function within complex biological systems, allowing researchers to study cellular responses, identify rare cell populations, and trace developmental lineages in a way that was not possible with bulk RNA-seq methods [2]. The fundamental difference between these approaches lies in whether each sequencing library reflects an individual cell or an averaged group of cells, with scRNA-seq requiring specialized methodologies to overcome challenges such as scarce transcripts, inefficient mRNA capture, and amplification biases [11]. This protocol provides a comprehensive overview of the complete scRNA-seq workflow, from sample preparation through data analysis, framed within the context of single-cell RNA sequencing protocol for isolates research.
scRNA-seq Workflow: A Step-by-Step Guide
Sample Preparation and Single-Cell Isolation
The initial and most critical step in conducting scRNA-seq is the effective isolation of viable, single cells from the tissue of interest. The quality of this starting material profoundly impacts all subsequent steps and the ultimate quality of the data [2]. For human bladder Ewing sarcoma tissue, as an example, this begins with generating highly viable single-cell suspensions while maintaining RNA integrity [9] [10]. The choice of isolation strategy depends on the tissue type, experimental goals, and available resources.
Table 1: Comparison of Major Single-Cell Isolation Methods
| Method | Principle | Advantages | Limitations | Throughput |
|---|---|---|---|---|
| FACS [11] | Fluorescent-activated cell sorting | High precision, ability to sort based on markers | Higher cost, potentially lower viability post-sorting | Medium |
| Droplet-based [12] | Microfluidic partitioning of cells | High-throughput, cost-effective per cell | Requires specialized equipment | High (thousands to millions of cells) |
| Microfluidic devices (e.g., Fluidigm C1) [2] | Microfluidic chip capture | Precise cell handling, integrated workflow | Lower throughput, higher cost per cell | Low to medium |
| Laser capture microdissection | Laser-based isolation | Spatial context preservation | Technically challenging, low throughput | Low |
| Split-pooling with combinatorial indexing [2] | Sequential barcoding in plates | Avoids specialized equipment, scalable to millions of cells | Complex workflow, lower efficiency | Very high |
Emerging methodologies such as single-nuclei RNA-seq (snRNA-seq) offer advantages when tissue dissociation is challenging, or when working with frozen or fragile samples [2] [11]. Similarly, 'split-pooling' scRNA-seq approaches based on combinatorial indexing of single cells allow for the processing of large sample sizes (up to millions of cells) without requiring expensive microfluidic hardware [2].
Figure 1: Sample Preparation and Single-Cell Isolation Workflow
Library Preparation and Sequencing
Once high-quality single-cell suspensions are obtained, the next critical phase involves preparing sequencing libraries. This process converts the minute amounts of RNA from individual cells into a format compatible with next-generation sequencing (NGS) platforms. The fundamental steps include cell lysis, reverse transcription, cDNA amplification, and the addition of sequencing adapters [2].
A groundbreaking innovation in this field has been the development of droplet-based partitioning systems, such as the 10x Genomics Chromium platform. This technology combines single cells, reverse transcription reagents, and barcoded Gel Beads with oil on a microfluidic chip to form reaction vesicles called GEMs (Gel Beads-in-emulsion). Each functional GEM contains a single cell, a single Gel Bead, and reverse transcription reagents. Within each GEM, the cell is lysed, the Gel Bead dissolves to release barcoded oligonucleotides, and reverse transcription of polyadenylated mRNA occurs [12]. Crucially, all cDNAs originating from the same cell share an identical barcode, enabling bioinformatic mapping of sequencing reads back to their cell of origin after sequencing [12].
Table 2: Comparison of scRNA-seq Protocols and Technologies
| Protocol | Isolation Strategy | Transcript Coverage | UMI | Amplification Method | Key Applications |
|---|---|---|---|---|---|
| 10x Genomics Chromium [12] | Droplet-based | 3'- or 5'-end counting | Yes | PCR | High-throughput cellular heterogeneity studies |
| Smart-Seq2 [11] | FACS | Full-length | No | PCR | Detection of low-abundance transcripts, isoform analysis |
| Drop-Seq [11] | Droplet-based | 3'-end | Yes | PCR | High-throughput, low cost per cell |
| CEL-Seq2 [11] | FACS | 3'-only | Yes | IVT | Reduced amplification bias |
| MATQ-Seq [11] | Droplet-based | Full-length | Yes | PCR | Accurate transcript quantification, variant detection |
| inDrop [11] | Droplet-based | 3'-end | Yes | IVT | Low cost per cell, efficient barcode capture |
The latest technological advancements include GEM-X technology, which generates twice as many GEMs at smaller volumes, reducing multiplet rates two-fold and increasing throughput capabilities. Modern platforms can process 80,000 to 960,000 cells per kit in a single instrument run with up to 80% cell recovery efficiency [12]. For enhanced sample compatibility, Flex assays enable profiling of fresh, frozen, and fixed samples, including FFPE tissues and fixed whole blood, providing crucial flexibility for clinical and longitudinal studies [12].
Figure 2: Library Preparation and Sequencing Workflow
Data Processing and Analysis
The transformation of raw sequencing data into biologically meaningful information requires a sophisticated computational workflow. After sequencing, the barcoded data must be processed to generate gene expression matrices and undergo multiple analytical steps to extract insights into cellular heterogeneity and function.
The initial computational processing typically involves tools like Cell Ranger (10x Genomics), which demultiplexes the sequencing data, aligns reads to a reference genome, and generates a feature-barcode matrix containing counts of RNA transcripts for each gene in each cell [12]. This raw count matrix, a numeric matrix of genes × cells, serves as the fundamental input for all subsequent analyses [13].
A critical preprocessing challenge arises from the nature of scRNA-seq data, which is inherently heteroskedastic—counts for highly expressed genes naturally vary more than for lowly expressed genes [13]. Various transformation approaches have been developed to address this issue:
- Delta method-based transformations: Include the shifted logarithm (e.g., log(y/s + y₀)) and the inverse hyperbolic sine (acosh) transformation, which aim to stabilize variance across the dynamic range of expression [13]
- Pearson residuals: Implemented in tools like sctransform, these residuals are calculated as (ygc - μ̂gc)/√(μ̂gc + α̂g μ̂_gc²) after fitting a gamma-Poisson generalized linear model [13]
- Latent expression inference: Methods like Sanity and Dino that infer parameters of generative models to estimate latent gene expression values [13]
- Count-based factor analysis: Approaches like GLM PCA and NewWave that directly model count distributions to produce low-dimensional representations [13]
Benchmark studies have shown that a rather simple approach—the logarithm with a pseudo-count followed by principal-component analysis—often performs as well or better than more sophisticated alternatives [13].
Figure 3: Data Processing and Analysis Workflow
Essential Reagents and Computational Tools
Research Reagent Solutions
Table 3: Essential Research Reagents for scRNA-seq Workflows
| Reagent/Category | Function | Examples/Notes |
|---|---|---|
| Cell Viability Kits | Assess viability of single-cell suspensions | Fluorescent live/dead stains, trypan blue; aim for >80% viability |
| Dissociation Enzymes | Tissue-specific digestion to single cells | Collagenase, trypsin, liberase; optimized for tissue type |
| Barcoded Gel Beads | Cell barcoding and mRNA capture | 10x Genomics Barcoded Gel Beads with oligo-dT primers |
| Reverse Transcription Mix | cDNA synthesis from captured mRNA | Includes reverse transcriptase, nucleotides, buffers |
| Partitioning Oil & Chips | Microfluidic formation of GEMs | 10x Genomics Partitioning Oil & Microfluidic Chips |
| Library Preparation Kits | Preparation of sequencing-ready libraries | Include enzymes for fragmentation, adapter ligation, and amplification |
| Cleanup Beads | Size selection and purification | SPRIselect, AMPure XP beads for cDNA and library cleanup |
| UMI Reagents | Unique Molecular Identifiers for digital counting | Molecular barcodes to distinguish biological from technical variation |
Computational Tools for scRNA-seq Analysis
The analysis of scRNA-seq data relies on a robust ecosystem of computational tools and pipelines. The choice of tools depends on the experimental design, computational resources, and biological questions being addressed.
- Processing Pipelines: Cell Ranger (10x Genomics) provides a comprehensive solution for processing raw sequencing data into gene-cell count matrices, performing secondary analysis including clustering, and generating preliminary visualizations [12]
- Quality Control: Tools like FastQC for sequencing quality assessment, and scRNA-seq specific metrics including genes per cell, UMIs per cell, and mitochondrial percentage
- Primary Analysis Platforms: Seurat and Scanpy represent two of the most widely used frameworks for comprehensive scRNA-seq analysis, providing functions for normalization, feature selection, dimensionality reduction, clustering, and differential expression [9] [13]
- Visualization Software: Loupe Browser (10x Genomics) enables interactive exploration of scRNA-seq data without requiring programming expertise [12]
- Specialized Applications: Monocle and Slingshot for trajectory inference; CellPhoneDB for cell-cell communication analysis; and SCENIC for gene regulatory network inference
For researchers seeking to contextualize their findings within existing knowledge, numerous public databases host scRNA-seq datasets, including GEO/SRA, Single Cell Expression Atlas (EMBL), Single Cell Portal (Broad Institute), CZ Cell x Gene Discover, and the scRNAseq package on Bioconductor, which provides curated datasets as SingleCellExperiment objects for easy integration with analysis pipelines [8].
The scRNA-seq workflow represents a transformative methodology for investigating cellular heterogeneity and function at unprecedented resolution. From careful sample preparation through sophisticated computational analysis, each step in the process contributes to the generation of high-quality data capable of revealing novel biological insights. As the technology continues to evolve, with improvements in throughput, sensitivity, and compatibility with diverse sample types, scRNA-seq is poised to drive further discoveries across biomedical research, drug discovery, and clinical applications. The integration of spatial transcriptomics methods, as demonstrated in bladder Ewing sarcoma protocols [9] [10], further enhances this powerful approach by adding crucial spatial context to single-cell gene expression profiles. For researchers embarking on scRNA-seq studies, attention to protocol details at each stage—from cell isolation to data transformation—is essential for obtaining reliable, interpretable results that advance our understanding of cellular biology in health and disease.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the dissection of gene expression at an unprecedented resolution, revealing cellular heterogeneity, identifying rare cell types, and illuminating developmental trajectories that are obscured in bulk sequencing approaches [1] [14]. The fundamental principle underlying all scRNA-seq technologies is the conversion of RNA from individual cells into cDNA libraries that can be sequenced, but the methodological approaches diverge significantly in how they capture transcript information [1] [15]. Since the first conceptual breakthrough in 2009, the field has rapidly evolved into two primary technological paradigms: full-length transcript protocols that sequence complete mRNA molecules, and 3' or 5' end counting protocols that focus on the terminal regions of transcripts [1] [15]. The choice between these pathways represents a critical decision point for researchers, with significant implications for experimental design, data quality, analytical possibilities, and resource allocation. This application note provides a comprehensive comparison of these approaches, offering structured guidance for researchers navigating this complex technological landscape within the context of a broader thesis on single-cell RNA sequencing protocols for isolates research.
Full-Length Transcript Sequencing Protocols
Full-length scRNA-seq methods are designed to capture and sequence complete mRNA molecules from the 5' cap to the 3' poly-A tail, preserving the entire transcript sequence information. These protocols typically employ oligo-dT priming to target the poly-A tail of mRNAs, followed by reverse transcription utilizing template-switching oligonucleotides (TSO) to capture the 5' end [16] [17]. The SMART-seq (Switching Mechanism at 5' End of RNA Template) technology and its subsequent iterations (SMART-seq2, SMART-seq3, SMART-seq HT) represent the most widely adopted implementations of this approach [17]. These methods generate sequencing libraries that cover the entire transcript length, enabling comprehensive characterization of transcriptional landscapes.
Key advantages of full-length protocols include their ability to detect alternative splicing events, single nucleotide variants, allele-specific expression, and comprehensive isoform diversity [16] [15]. Methods like RamDA-seq have further expanded these capabilities to include total RNA sequencing, capturing both polyadenylated and non-polyadenylated RNAs, including nascent transcripts, histone mRNAs, long noncoding RNAs, and enhancer RNAs [16]. This comprehensive coverage comes with specific technical requirements, including higher sequencing depth per cell to adequately cover full-length transcripts, which typically results in higher costs per cell and lower overall throughput compared to end-counting methods [17] [15].
3'/5' End Counting Protocols
End counting methods represent a fundamentally different approach that focuses sequencing efforts on either the 3' or 5' ends of transcripts, utilizing unique molecular identifiers (UMIs) for precise transcript quantification [1] [18]. These protocols typically employ droplet-based microfluidics systems (e.g., 10x Genomics Chromium, Drop-seq, inDrop) or combinatorial indexing strategies (e.g., FIPRESCI) to process thousands to hundreds of thousands of cells in a single experiment [1] [19]. The core innovation lies in the incorporation of cell barcodes and UMIs during reverse transcription, enabling massive multiplexing and accurate digital counting of transcript molecules.
The 5' end methods (e.g., STRT-seq, FB5P-seq, FIPRESCI) capture the beginning of transcripts, providing advantages for identifying transcription start sites, promoter usage, and enhancer activity [18] [19]. Additionally, 5' end protocols are particularly suited for immune repertoire sequencing as they naturally cover the variable regions of T-cell receptors (TCR) and B-cell receptors (BCR) [18] [19]. In contrast, 3' end methods (e.g., Drop-seq, Chromium, Seq-Well) focus on the transcript termini, providing robust gene expression quantification with high cell throughput [15]. Both approaches sacrifice complete transcript information for dramatically increased scalability and reduced per-cell costs, making them ideal for large-scale atlas projects and studies focusing on cellular heterogeneity rather than isoform diversity.
Visual Comparison of Core Methodologies
The following workflow diagrams illustrate the fundamental technical differences between full-length and end-counting approaches, highlighting key branching points in experimental design.
Comparative Analysis: Technical Specifications and Performance Metrics
Direct comparison of full-length versus end-counting protocols requires evaluation across multiple technical parameters. The following tables provide a structured analysis of key characteristics, performance metrics, and cost considerations to inform experimental design decisions.
Technical Specifications and Applications
Table 1: Core Technical Characteristics of Major scRNA-seq Protocol Types
| Parameter | Full-Length Protocols | 3' End Counting | 5' End Counting |
|---|---|---|---|
| Transcript Coverage | Complete mRNA molecule (5' to 3') | 3' terminal region only | 5' start region only |
| Priming Strategy | Oligo-dT + template switching | Oligo-dT with barcodes | Oligo-dT or random with barcodes |
| UMI Incorporation | Limited implementations (e.g., SMART-seq3) | Standard feature | Standard feature |
| Throughput (Cells) | Low to medium (96-1,000 cells) | High (10,000-100,000 cells) | High (10,000-100,000 cells) |
| Sensitivity (Genes/Cell) | High (7,000-10,000 genes) | Medium (1,000-5,000 genes) | Medium (1,000-5,000 genes) |
| Key Applications | Alternative splicing, mutation detection, isoform discovery | Cell typing, trajectory inference, differential expression | Immune repertoire, transcription start sites, enhancer activity |
| Representative Methods | SMART-seq2, SMART-seq3, RamDA-seq, MATQ-seq | 10x Genomics Chromium, Drop-seq, Seq-Well | STRT-seq, FB5P-seq, FIPRESCI |
Performance Benchmarking and Cost Analysis
Table 2: Performance and Practical Considerations for Selected Protocols
| Protocol | Type | Genes/Cell | Cells/Run | Cost/Cell (€) | Strengths | Limitations |
|---|---|---|---|---|---|---|
| SMART-seq3 | Full-length | ~10,000 | 96-384 | ~12 | High sensitivity, UMIs, isoform information | Lower throughput, higher hands-on time |
| G&T-seq | Full-length | ~9,500 | 96 | ~12 | Parallel genome & transcriptome, high sensitivity | Complex workflow, low throughput |
| Takara SMART-seq HT | Full-length | ~8,000 | 96-384 | ~73 | Automation friendly, consistent performance | Highest cost, medium sensitivity |
| 10x Chromium (3') | 3' end | 1,000-5,000 | 10,000 | ~0.70 | High throughput, standardized, easy use | Limited transcript information |
| FB5P-seq | 5' end | 2,000-5,000 | 96-384 | ~2.70 | Immune repertoire, FACS integration | Medium throughput, specialized application |
| FIPRESCI | 5' end | 1,000-4,000 | 100,000+ | <0.50 | Massive scalability, low multiplet rate | Complex barcoding, newer method |
Detailed Methodologies: Protocol Implementation
Full-Length Protocol: SMART-seq3 Workflow
SMART-seq3 represents the cutting edge of full-length scRNA-seq protocols, incorporating UMIs while maintaining full-length transcript coverage [17]. The detailed experimental procedure encompasses the following critical stages:
Single-Cell Isolation and Lysis: Individual cells are sorted into 96- or 384-well plates containing lysis buffer using FACS or microfluidic platforms. The lysis buffer typically includes detergents (e.g., Triton X-100), RNase inhibitors, dNTPs, and oligo-dT primers. Each well receives a single cell, and plates are immediately frozen on dry ice and stored at -80°C until processing.
Reverse Transcription and Template Switching: Reverse transcription is performed using Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase with template-switching activity. The reaction includes:
- Template Switching Oligo (TSO) with unique molecular identifiers (UMIs)
- Oligo-dT primer containing well-specific barcodes
- Betaine and MgCl₂ to enhance reaction efficiency
- Incubation: 90 min at 42°C, followed by enzyme inactivation at 70°C
cDNA Amplification and Purification: Full-length cDNA is amplified via PCR (18-22 cycles) using ISPCR primers. The amplification reaction utilizes a high-fidelity DNA polymerase with proofreading activity. Amplified cDNA is purified using solid-phase reversible immobilization (SPRI) beads with a 0.6-0.8× bead-to-sample ratio to remove primers, enzymes, and short fragments.
Library Preparation and Sequencing: Tagmentation-based library preparation (e.g., Nextera XT) fragments the cDNA and adds sequencing adapters. Library quality is assessed using capillary electrophoresis (e.g., Bioanalyzer), and quantification is performed via fluorometry. Sequencing is typically performed on Illumina platforms with 2×150 bp paired-end reads to ensure complete transcript coverage.
End-Counting Protocol: FIPRESCI Workflow for 5' End Sequencing
FIPRESCI (FIve PRime End Single-cell Combinatorial Indexing) combines droplet microfluidics with combinatorial indexing to achieve massive throughput for 5' end sequencing [19]. The protocol involves these key steps:
Cell Permeabilization and Reverse Transcription: Cells or nuclei are fixed and permeabilized to allow reagent access while maintaining structural integrity. Reverse transcription is performed using oligo-dT primers or random hexamers in suspension. For nuclei preparations, this step captures nuclear transcripts including nascent RNA.
Combinatorial Pre-Indexing with Tn5 Transposase: Permeabilized cells/nuclei are distributed across a 96-well plate, with each well containing Tn5 transposase loaded with unique barcoded oligonucleotides. The Tn5 simultaneously fragments and tags cDNA molecules with well-specific barcodes (Round 1 barcoding). Critically, the reaction occurs without SDS to maintain Tn5 binding to cDNA.
Droplet-Based Barcoding with High Overloading: Pre-indexed cells are pooled and loaded into a microfluidic droplet system (e.g., 10x Genomics Chromium) at high concentrations (10-20× standard loading). Within droplets, cDNA undergoes template switching with TSO containing:
- Droplet-specific barcodes (Round 2 barcoding)
- Unique Molecular Identifiers (UMIs)
- PCR handle sequences
Library Amplification and Target Enrichment: Droplets are broken, and barcoded cDNA is pooled and amplified via PCR (12-14 cycles). For immune repertoire analysis, an additional targeted PCR enrichment is performed using constant region primers for TCR or BCR sequences. Final libraries are quantified and sequenced with a custom read structure to decode combinatorial indices.
Research Reagent Solutions: Essential Materials and Tools
Table 3: Key Research Reagents and Their Applications in scRNA-seq Protocols
| Reagent Category | Specific Examples | Function | Protocol Applicability |
|---|---|---|---|
| Reverse Transcriptases | M-MLV RT, SmartScribe | cDNA synthesis with template switching | Universal, critical for full-length |
| Template Switching Oligos | TSO with LNA modifications, TSO with UMIs | 5' cDNA capture, enhances efficiency | Full-length protocols, some 5' end |
| Barcoding Systems | 10x Barcoded Gel Beads, Custom Tn5 | Cell and molecular indexing | End-counting methods, combinatorial indexing |
| Amplification Kits | NEBNext Ultra II FS, KAPA HiFi | cDNA amplification, library preparation | Universal with protocol-specific optimization |
| Library Prep Kits | Nextera XT, Illumina TruSeq | Sequencing library generation | Method-dependent selection |
| Cell Viability Assays | Propidium iodide, DAPI, Calcein AM | Assessment of cell integrity pre-processing | Universal quality control |
| mRNA Capture Beads | Oligo-dT Magnetic Beads | mRNA purification from lysate | G&T-seq, other purification-needed protocols |
Application Contexts: Matching Protocols to Biological Questions
Guidance for Protocol Selection
The optimal scRNA-seq protocol depends fundamentally on the specific research question and experimental constraints. The following decision framework provides guidance for selecting the most appropriate methodology:
Choose Full-Length Protocols When: Investigating alternative splicing patterns in cancer subtypes [15], characterizing isoform diversity in neuronal development [16], identifying allele-specific expression in heterogeneous tissues [15], or studying non-polyadenylated RNAs (e.g., enhancer RNAs, histone mRNAs) using specialized methods like RamDA-seq [16].
Choose 3' End Counting When: Constructing cell atlases of complex tissues (e.g., human cell atlas projects) [1], analyzing cellular heterogeneity in tumor microenvironments [14], performing large-scale drug screening with single-cell resolution [14], or tracing developmental trajectories with high cell numbers [15].
Choose 5' End Counting When: Studying adaptive immune responses with paired TCR/BCR sequencing [18] [19], investigating transcription start site usage and promoter activity [19], analyzing cis-regulatory elements and enhancer activity [19], or requiring high-throughput analysis with immune receptor profiling.
Emerging Applications and Future Directions
Recent methodological advances continue to expand scRNA-seq applications. The integration of spatial transcriptomics with single-cell approaches addresses the limitation of lost spatial context in conventional scRNA-seq [14]. Multi-omics technologies like G&T-seq enable parallel analysis of genome and transcriptome from the same cell, providing opportunities to connect genotypes with transcriptional phenotypes [17]. Combinatorial indexing strategies like FIPRESCI push throughput boundaries, enabling massive-scale experiments approaching millions of cells [19]. Additionally, long-read single-cell sequencing is emerging as a powerful approach for direct isoform characterization without inference, as demonstrated in bulk Iso-Seq studies of immune cells [20].
The strategic selection between full-length and end-counting scRNA-seq protocols represents a fundamental decision point in experimental design, with significant implications for data quality, analytical capabilities, and resource allocation. Full-length methods provide comprehensive transcript information essential for isoform-level analyses but at higher per-cell costs and lower throughput. Conversely, end-counting approaches offer unprecedented scalability for cellular heterogeneity studies while sacrificing detailed transcript structure information. As the field continues to evolve, emerging technologies like combinatorial indexing and long-read single-cell sequencing promise to further expand these capabilities. By understanding the technical foundations, performance characteristics, and application fit of these distinct approaches, researchers can make informed decisions that align methodological capabilities with biological questions, ultimately maximizing the scientific return on investment in single-cell transcriptomics.
The Role of Unique Molecular Identifiers (UMIs) and Spike-Ins in Quantitative Accuracy
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical research by enabling the characterization of transcriptomes at the resolution of individual cells, revealing cellular heterogeneity that is obscured in bulk RNA sequencing approaches [21] [2]. Despite its transformative potential, a significant challenge in scRNA-seq lies in achieving precise quantitative accuracy in transcript measurement due to multiple technical variables including amplification biases, molecular capture efficiency, and sequencing artifacts [22] [23]. The minute starting amounts of RNA in individual cells (typically 10⁵–10⁶ mRNA molecules) necessitate substantial amplification, which introduces quantitative distortions that can compromise data integrity [24] [2].
To address these challenges, two powerful methodological approaches have been developed: Unique Molecular Identifiers (UMIs) and spike-in controls. UMIs are short, random nucleotide sequences (typically 4-12 bases) that are incorporated during reverse transcription to uniquely tag individual mRNA molecules, enabling bioinformatic correction for amplification biases [24] [1]. Spike-in controls are synthetic RNA molecules added in known quantities to samples, serving as internal standards for quality control and normalization [23] [25]. When implemented together within a scRNA-seq workflow, these tools provide a robust framework for achieving unprecedented quantitative accuracy in single-cell transcriptomics, which is particularly crucial for applications in drug development and precision medicine where reliable quantification of subtle transcriptional changes is essential [26].
Fundamental Principles of UMIs
Technical Implementation and Mechanism of Action
Unique Molecular Identifiers function as molecular barcodes that are attached to each cDNA molecule during the initial reverse transcription step, prior to any amplification [24]. The core principle is that all PCR amplicons derived from a single original mRNA molecule will share an identical UMI sequence. Following sequencing, bioinformatics pipelines can collapse reads with identical UMIs and mapping coordinates into a single count, thereby eliminating artifactual inflation from PCR duplicates [24] [25]. This process, known as deduplication, ensures that the final count for each transcript reflects the true number of original molecules rather than amplification artifacts.
The effectiveness of UMI-based counting depends on several critical parameters. The length and complexity of the UMI sequence directly influences the maximum number of molecules that can be uniquely tagged. For instance, a 10-nucleotide UMI can theoretically generate 4¹⁰ (1,048,576) unique combinations, which is generally sufficient for tagging the approximately 100,000-1,000,000 mRNA molecules typically found in a mammalian cell [24]. However, UMI sequences are susceptible to errors during PCR amplification and sequencing, necessitating computational error correction strategies that typically involve collapsing UMIs within a certain Hamming distance (e.g., 1-2 nucleotides) of each other [22].
Practical Considerations for UMI Design and Implementation
The quantitative performance of UMIs is significantly influenced by experimental design choices. A comprehensive study evaluating UMI-based scRNA-seq protocols revealed that UMI lengths of at least 8 nucleotides are necessary for accurate RNA counting across a wide range of expression levels, with shorter UMIs exhibiting elevated collision rates that lead to undercounting [22]. Furthermore, specific protocol steps can dramatically impact counting accuracy. For example, in tSCRB-seq protocol implementations that omit cleanup steps after reverse transcription, residual primers can cause substantial UMI overcounting, linearly correlating with sequencing depth rather than true biological expression [22].
Table 1: Impact of UMI Length on Quantitative Accuracy
| UMI Length | Theoretical Diversity | Molecular Exponent* | Counting Accuracy | Primary Limitation |
|---|---|---|---|---|
| 4 nt | 256 | ~0.6 | Low | Severe undercounting due to limited diversity |
| 6 nt | 4,096 | <0.8 | Moderate | Elevated collision rates |
| 8 nt | 65,536 | ~0.8 | Good | Accurate for most expression levels |
| 10 nt | 1,048,576 | ~0.8-1.0 | Excellent | Optimal for full expression range |
| 12 nt | 16,777,216 | ~1.0 | Excellent | Minimal collisions, higher sequencing cost |
The molecular exponent describes the relationship between input molecules and UMI counts (ideal value = 1) [23]
Spike-In Controls: Types and Applications
Categories of Spike-In Controls
Spike-in controls can be broadly categorized into two main types: synthetic RNA spike-ins and cellular spike-ins. The most widely used synthetic spike-ins are the External RNA Controls Consortium (ERCC) standards, which comprise 92 synthetic RNA transcripts with varying lengths and GC content, mixed at known concentrations that span a wide dynamic range [23] [25]. These are typically added to cell lysates at the beginning of library preparation. More recently, "molecular spikes" have been developed that incorporate built-in UMIs, creating an experimental ground truth that enables direct assessment of a protocol's RNA counting performance [22] [27].
Cellular spike-ins involve adding well-characterized cells (e.g., mouse 32D cells into human samples) prior to processing, creating internal controls that enable detection and correction for sample-specific contamination. This approach has proven particularly valuable for identifying cross-contamination from cell-free RNA, which can constitute up to 20% of reads in primary tissue samples [26].
Implementation Framework for Spike-In Controls
The effective use of spike-in controls requires careful experimental planning. For synthetic RNA spike-ins like ERCC standards, they should be added at the earliest possible stage of library preparation—ideally during cell lysis—to subject them to the entire experimental workflow alongside endogenous transcripts [23]. The spike-in molecules should cover the entire expected dynamic range of endogenous expression, with concentrations calibrated to match the expected RNA content of the cells being studied [23].
For cellular spike-ins, the reference cells should be fixed to prevent any transcriptional responses during processing, and typically comprise 5-10% of the total cell population [26]. Cross-species spike-ins (e.g., mouse cells in human samples) are particularly advantageous as they allow unambiguous separation of spike-in and sample-derived transcripts during bioinformatic analysis through alignment to separate reference genomes [26].
Diagram 1: Spike-in implementation workflow illustrating parallel paths for synthetic and cellular spike-ins
Integrated Experimental Protocols
Protocol 1: Molecular Spikes for scRNA-seq Validation
Molecular spikes with built-in UMIs provide a robust method for validating the quantitative performance of scRNA-seq protocols [22] [27]. The following protocol outlines their implementation:
Reagents Required:
- Molecular spike RNA pools (5′ or 3′ molecular spikes with 18-nt spUMIs)
- scRNA-seq library preparation kit (e.g., Smart-seq3, 10x Genomics)
- Standard cell culture reagents for the chosen cell line
Procedure:
- Spike-in Calibration: Dilute molecular spikes to appropriate concentrations spanning the expected expression range of endogenous transcripts (typically 1-1,000 molecules per cell).
- Sample Preparation: Add calibrated molecular spikes to single-cell suspensions of HEK293FT cells or other relevant cell lines.
- Library Preparation: Process samples according to standard scRNA-seq protocols (e.g., Smart-seq3 for full-length coverage or 10x Genomics for 3′-tagging).
- Sequencing: Sequence libraries following platform-specific recommendations.
- Bioinformatic Analysis:
- Extract spUMI sequences from aligned reads
- Apply error correction using a Hamming distance of 2 nucleotides
- Exclude over-represented spUMIs across cells to remove potential biases
- Compare observed spUMI counts to expected values to calculate counting accuracy
Validation Metrics: The protocol should yield a strong correlation (r² > 0.99) between observed error-corrected counts and the known number of spiked molecules across the tested concentration range [22].
Protocol 2: Cellular Spike-ins for Contamination Detection
This protocol utilizes cross-species cellular spike-ins to identify and correct for RNA contamination in scRNA-seq experiments [26]:
Reagents Required:
- Mouse 32D cells (for human samples) or other cross-species cell lines
- Methanol for fixation
- Single-cell suspension of primary tissue (e.g., human pancreatic islets)
- 10X Chromium platform or similar droplet-based system
Procedure:
- Spike-in Cell Preparation: Fix approximately 5×10⁵ mouse 32D cells in methanol and store at -80°C until use.
- Sample Preparation: Dissociate primary human tissue (e.g., pancreatic islets) to single-cell suspension and determine viable cell count.
- Spike-in Addition: Thaw fixed mouse cells and add to human cell suspension at a ratio of 1:20 (mouse:human cells).
- Single-Cell Processing: Immediately load the mixed cell suspension onto the 10X Chromium controller following manufacturer's instructions.
- Library Preparation and Sequencing: Proceed with standard 10X Genomics library preparation and sequencing.
- Bioinformatic Analysis:
- Align sequences to a combined human-mouse reference genome
- Separate cells by species origin using the ratio of human:mouse reads
- Quantify cross-species contamination by measuring human reads in mouse cells
- Apply computational decontamination algorithms to correct endogenous cell expression profiles
Quality Assessment: Effective implementation typically reduces contamination artifacts from >15% to <2% of reads, significantly improving detection of true biological signals, particularly for low-abundance transcripts [26].
Table 2: Performance Comparison of scRNA-seq Protocols with Spike-in Controls
| scRNA-seq Protocol | Sensitivity (50% Detection Probability) | Accuracy (Pearson Correlation with Spike-ins) | UMI Efficiency | Best Application Context |
|---|---|---|---|---|
| SMART-seq2 | ~10 molecules | 0.85-0.95 | N/A (Full-length) | Alternative splicing, mutation detection |
| 10X Genomics (3′) | ~100 molecules | 0.80-0.90 | ~80% | High-throughput cell atlas construction |
| CEL-seq2 | ~10 molecules | 0.75-0.85 | ~75% | Plate-based targeted studies |
| MARS-seq | ~50 molecules | 0.70-0.80 | ~70% | High-throughput screening |
| Drop-seq | ~100 molecules | 0.75-0.85 | ~75% | Cost-effective large-scale studies |
| inDrop | ~10 molecules | 0.75-0.85 | ~75% | High-sensitivity applications |
Research Reagent Solutions
Table 3: Essential Research Reagents for UMI and Spike-in Applications
| Reagent Category | Specific Examples | Function | Implementation Considerations |
|---|---|---|---|
| Synthetic RNA Spike-ins | ERCC RNA Spike-In Mix (Thermo Fisher) | Protocol normalization and sensitivity assessment | Covers 6 logs of dynamic range; 92 transcripts |
| Molecular Spikes | 5′ and 3′ molecular spikes with spUMIs [22] | Direct assessment of RNA counting accuracy | Built-in 18-nt UMIs; enables ground-truth validation |
| Cellular Spike-ins | Mouse 32D cells, Human Jurkat cells [26] | Contamination detection and correction | Cross-species application recommended |
| UMI Library Prep Kits | 10X Genomics Chromium, SMART-seq3, CEL-seq2 | Incorporation of UMIs during cDNA synthesis | Varying UMI lengths (6-12 nt) impact counting accuracy |
| Bioinformatics Tools | UMI-tools, zUMIs, UMIcountR [22] [23] | Demultiplexing, error correction, and deduplication | Hamming distance parameters must be optimized |
| Reference Transcriptomes | Combined species references (e.g., human-mouse) | Bioinformatic separation of spike-in and sample reads | Essential for cellular spike-in analysis |
Data Analysis Framework
Bioinformatics Processing Pipeline
The analysis of UMI and spike-in scRNA-seq data requires specialized bioinformatic processing to extract quantitative insights. A standardized workflow encompasses multiple stages:
Primary Processing:
- Demultiplexing: Separate sequencing data by sample barcodes
- UMI Extraction: Identify and record UMI sequences from read headers or sequences
- Alignment: Map reads to an appropriate reference genome (possibly combined species for cellular spike-ins)
- Deduplication: Collapse reads with identical UMIs and mapping coordinates
Spike-in Specific Processing:
- Spike-in Identification: Separate endogenous and spike-in reads using predefined identifiers
- Contamination Assessment: For cellular spike-ins, quantify cross-species alignment rates
- Normalization: Use spike-in derived factors to adjust for technical variability between samples
- Error Correction: Apply UMI error correction using hierarchical clustering or network-based approaches
Diagram 2: Bioinformatic workflow for processing UMI and spike-in scRNA-seq data
Normalization Strategies Using Spike-ins
Spike-in controls enable sophisticated normalization approaches that account for technical variability in capture and amplification efficiency. The most effective methods include:
Spike-in Based Size Factors: Calculate size factors for each cell based on spike-in counts to normalize for technical differences in capture efficiency, applying the formula:
SF_cell = median(spike-in counts_cell / mean spike-in counts across all cells)
Multipath Normalization: Combine spike-in derived factors with endogenous content factors to address both capture efficiency and sequencing depth variations.
Contamination Correction: For cellular spike-ins, use the contamination profile observed in spike-in cells to computationally remove background contamination from endogenous cell expression data [26].
These normalization strategies have been shown to significantly improve quantitative accuracy, particularly when analyzing subtle transcriptional changes in drug response studies or identifying rare cell populations [26].
Applications in Drug Development and Biomedical Research
The implementation of UMI and spike-in controls has enabled particularly impactful applications in pharmaceutical research and development, where quantitative accuracy is paramount for reliable decision-making.
In drug mechanism-of-action studies, researchers have employed spike-in controlled scRNA-seq to elucidate cell-type-specific drug effects in complex tissues. For example, when applying this approach to human and mouse pancreatic islets treated with various drugs including artemether and a FOXO inhibitor, researchers obtained precise quantification of cell-specific drug effects, observing that FOXO inhibition induced dedifferentiation of both alpha and beta cells [26]. This level of quantitative resolution enables more confident identification of cell-type-specific responses and potential off-target effects during preclinical drug evaluation.
In oncology applications, UMI-enhanced scRNA-seq has proven invaluable for characterizing tumor heterogeneity and identifying rare drug-resistant subpopulations that might be missed without rigorous quantitative controls. The ability to accurately count transcripts using UMIs allows researchers to distinguish true biological variation from technical artifacts, enabling more reliable identification of biomarkers and drug targets [2]. Furthermore, cellular spike-in approaches have been instrumental in detecting and correcting for ambient RNA contamination in tumor microenvironments, where high levels of cell death can significantly compromise data quality [26].
The integration of these quantitative controls has also accelerated the development of cell atlases in regenerative medicine, providing reference standards for cellular composition and expression levels that enable robust comparison across studies, laboratories, and experimental conditions [1] [21]. As single-cell technologies continue to evolve toward clinical applications, UMI and spike-in methodologies will play an increasingly critical role in ensuring the reproducibility and reliability of results that inform therapeutic development.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the examination of gene expression patterns at the ultimate level of resolution: the individual cell. This technology has become the state-of-the-art approach for unravelling the heterogeneity and complexity of RNA transcripts within individual cells, as well as revealing the composition of different cell types and functions within highly organized tissues/organs/organisms [1]. Since its conceptual breakthrough in 2009, scRNA-seq has provided massive information across different fields, leading to exciting new discoveries in understanding cellular composition and interactions [1]. The ability to profile gene expression activity in individual cells represents one of the most authentic approaches to probe cell identity, state, function, and response, allowing researchers to classify, characterize, and distinguish each cell at the transcriptome level [1]. This technical capability has opened two particularly powerful application domains: the discovery and characterization of rare cell populations that are functionally important but often missed in bulk analyses, and the tracing of lineage trajectories that reveal developmental pathways and cellular differentiation processes. These applications are transforming our understanding of cellular heterogeneity, disease mechanisms, and developmental biology, providing unprecedented insights into the complex organization of biological systems from humans to model animals and plants [1].
Discovering Rare Cell Types
Technological Foundation and Principles
The power of scRNA-seq to identify rare cell populations stems from its ability to analyze transcriptomes at single-cell resolution for over millions of cells in a single study [1]. Unlike traditional bulk RNA sequencing, which measures average gene expression across thousands or millions of cells, thereby obscuring rare cellular signatures, scRNA-seq captures the unique transcriptional profile of each individual cell. This capability is particularly valuable for identifying rare but functionally critical cell types that exist in low frequencies within tissues, such as stem cells, progenitor cells, or rare pathological cells [1]. The technological foundation for this application involves several key steps: single-cell isolation and capture, cell lysis, reverse transcription (conversion of RNA into cDNA), cDNA amplification, and library preparation [1]. The dramatic reduction in cost and significant increase in automation and throughput have made large-scale scRNA-seq studies feasible, enabling comprehensive cataloging of cellular diversity [1]. The introduction of unique molecular identifiers (UMIs) has further enhanced the quantitative nature of scRNA-seq by effectively eliminating PCR amplification bias, thereby improving the accuracy of rare cell population identification [1].
Experimental Protocol for Rare Cell Discovery
Sample Preparation and Single-Cell Suspension: The initial critical step involves generating high-quality single-cell suspensions from the tissue of interest. For live cell dissociations, this typically includes a combination of enzymatic digestion of extracellular components and mechanical disruption to separate cells while maintaining viability [28]. The dissociation process must be performed rapidly to minimize transcriptomic responses to dissociation stress. Performing digestions on ice can help mediate these transcriptional responses, though this may slow digestion times as most commercially available enzymes are optimized for activity at 37°C [28]. Recently, fixation-based methods have been applied to relieve some of these issues by essentially stopping the transcriptomic response. Methods include methanol maceration optimized for single cell sequencing (ACME) or reversible dithio-bis(succinimidyl propionate) (DSP) fixation immediately following cell dissociation [28]. For tissues that are difficult to dissociate, single-nucleus RNA sequencing (snRNA-seq) provides an alternative approach that solves problems related to tissue preservation and cell isolation [1].
Cell Capture and Library Preparation: Current commercially available solutions for cell capture and library generation vary in their mechanisms and capacity. The 10x Genomics platform offers a droplet microfluidics solution with the flexibility to capture as few as 500 or as many as 20,000 cells with their latest GEM-X v4 assay [28]. Illumina provides a vortex-based single cell droplet capture solution that can process a wide range of inputs without microfluidics platform restrictions [28]. Alternative approaches include sorting cells into microwells (BD Rhapsody, Singleron) with larger maximal size capacity than microfluidics approaches, and plate-based combinatorial barcoding solutions (Scale BioScience, Parse BioScience) that can return over 100,000 cells [28]. The choice of platform depends on the specific research requirements, including the number of cells needed, cell size characteristics, and project scale.
Bioinformatic Analysis and Clustering: Following sequencing, bioinformatic analysis is performed to identify rare cell populations. This process involves mapping reads to an adequate reference to generate a count matrix, followed by downstream analyses of expression profiles [28]. Clustering analysis has become an essential tool for interpreting and revealing cell types and functional states within these complex datasets [29]. Advanced clustering methods such as Seurat, PhenoGraph, SC3, and Scanpy are employed to identify groups of cells with similar expression patterns [30] [29]. The recently developed scMSCF (single-cell Multi-Scale Clustering Framework) introduces a multi-dimensional PCA strategy for dimensionality reduction, combined with K-means clustering and a weighted ensemble meta-clustering approach enhanced by a self-attention-driven Transformer model to optimize clustering performance [29]. This method has demonstrated improved ability to identify diverse cell populations, achieving on average 10-15% higher ARI, NMI, and ACC scores compared to existing methods [29].
Key Applications and Impact
The discovery of rare cell types through scRNA-seq has profound implications across multiple research domains. In developmental biology, scRNA-seq has enabled the identification of rare progenitor populations during organogenesis and tissue formation [1]. In cancer research, the technology has revealed rare tumor-initiating cells and resistance-conferring subpopulations within heterogeneous tumors [1]. In neuroscience, scRNA-seq has uncovered specialized neuronal subtypes that were previously unrecognized [1]. The application of scRNA-seq to build comprehensive cell atlases for various organisms and tissues represents one of the most significant contributions of this technology, serving as key resources for better understanding and treating diseases [1]. These atlases provide high-resolution catalogs of cells in living organisms, enabling researchers to reference rare cell populations across different conditions and disease states.
Table 1: scRNA-seq Clustering Methods for Rare Cell Type Identification
| Method | Key Features | Advantages for Rare Cell Discovery | References |
|---|---|---|---|
| Seurat | Integrates normalization, scaling, PCA; constructs cell similarity graph via k-nearest neighbors | Effective community detection; widely validated | [30] [29] |
| PhenoGraph | Constructs cell relationships using k-nearest neighbors; applies community detection algorithms | Suitable for biologically related cell types with subtle expression differences | [29] |
| SC3 | Utilizes multiple clustering results to enhance clustering decisions | Improved consensus approach for robust clustering | [29] |
| scMSCF | Combines multi-dimensional PCA, K-means, weighted ensemble meta-clustering, and Transformer model | 10-15% higher ARI, NMI, and ACC scores; captures complex dependencies | [29] |
| SHARP | Ensemble strategies based on random projections | Improved clustering speed and accuracy for large, noisy datasets | [29] |
Tracing Lineage Trajectories
Fundamental Concepts and Historical Development
Lineage tracing represents an essential approach for understanding cell fate, tissue formation, and human development [31]. At its core, lineage tracing encompasses any experimental design aimed at establishing hierarchical relationships between cells, enabling researchers to investigate cell morphology, differentiation, clonal expansion, and gene inheritance/expression [31]. The resolution and methodological approach define the limits of an analysis, balancing precision and generalizability. The field has evolved significantly from its origins in the late 1800s, when Charles Whitman reported the direct observation of germ layer differentiation in leeches [31]. The introduction of labeling technologies represented a major advancement, beginning with Eric Vogt's 1929 fate mapping of an amphibian blastula using Nile Blue [31]. The late 20th century witnessed exponential development of gene editing technologies, particularly through the manipulation of reporter genes that circumvented limitations of earlier approaches [31]. The introduction of green fluorescent protein (GFP) as an endogenous reporter in 1994 marked a massive shift for lineage tracing, giving cells the potential to express reporters without the need for an external stimulus [31]. These historical developments laid the foundation for modern lineage tracing approaches that integrate sophisticated imaging with scRNA-seq technologies.
Imaging-Based Lineage Tracing Techniques
Site-Specific Recombinase Systems: Central to imaging-based lineage-tracing research are site-specific recombinase (SSR) systems, with Cre-loxP remaining one of the most fundamental and commonly used [31]. These systems can knock-in/knock-out alleles and influence gene expression with a great degree of cell and temporal specificity. In lineage tracing, Cre-loxP systems are commonly applied in clonal analysis studies, where Cre recombinase excises a STOP codon between two adjacent loxP binding sites, activating a fluorescent reporter gene [31]. The specificity of this activation depends on Cre, whose expression can be driven by cell-type-specific promoters or ubiquitously expressed. A significant limitation of single fluorescent reporters is their difficulty in resolving cell populations at the single-cell level due to the challenge of distinguishing clonal groups within homogenously labelled populations [31]. Sparse labelling approaches can circumvent this by titrating the activating agent in inducible models to limit recombination to a limited number of cells within the population, thus allowing for spatial separation [31].
Dual Recombinase Systems: The Cre-loxP system can be combined with analogous technologies like Dre-rox to create dual recombinase systems [31]. These systems take advantage of the site specificity of recombinases and offer multiple experimental design strategies beneficial to lineage tracing, including expression occurring following recombination of either Cre or Dre, both Cre and Dre, or Cre in the absence of Dre [31]. Dual systems have been successfully applied to determine the origin of regenerative cells in remodelled bone, investigate cellular origins of alveolar epithelial stem cells post-injury, and develop novel genetic techniques for evaluating senescence [31]. For example, a Cre/Dre dual system was used to distinguish otherwise homogenous periosteal tissue into distinct layers and evaluate layer contributions in fracture regeneration [31].
Multicolour Lineage Tracing Approaches: A major advance in imaging-based lineage tracing was the introduction of multicolour reporter cassettes, beginning with "Brainbow," capable of expressing up to four different fluorescent proteins driven by stochastic Cre-loxP-mediated excision and/or inversion [31]. This technology has since been adapted to several experimental models and inspired numerous analogous technologies [31]. The R26R-Confetti reporter represents one of the most popular adaptations due to its widespread applicability to existing Cre models [31]. Lineage-tracing studies now incorporate confetti reporters for clonal analysis at the single-cell level in various tissues, including hematopoietic, epithelial, kidney, and skeletal cells [31]. Multicolour models are also being applied in live-imaging studies; for example, confetti reporters have recently been used in intravital imaging to trace macrophage origin and proliferation in mammary glands in real time [31].
Table 2: Lineage Tracing Techniques and Their Applications
| Technique | Mechanism | Resolution | Key Applications |
|---|---|---|---|
| Cre-loxP System | Site-specific recombination activates reporter expression | Population to single-cell (with sparse labelling) | Broad applicability; clonal analysis; inducible systems |
| Dual Recombinase Systems | Combined recombinase systems (e.g., Cre-loxP/Dre-rox) | Enhanced cellular resolution | Distinguishing homogeneous tissue layers; simultaneous tracking of multiple populations |
| Multicolour Reporters (Brainbow, Confetti) | Stochastic recombination generates multiple fluorescent hues | Single-cell | Clonal analysis in hematopoetic, epithelial, kidney, skeletal cells; intravital imaging |
| MADM (Mosaic Analysis with Double Markers) | Sparse labelling with two markers | Single-cell | Developmental lineage tracing; sparse tissue analysis |
| Nucleoside Analogues (BrdU, EdU) | Incorporation into cellular DNA during proliferation | Population level | Identifying proliferating cell populations; label dilution indicates proliferation history |
Integration with scRNA-seq for Lineage Tracing
The integration of lineage tracing with scRNA-seq represents a powerful approach that combines the strengths of both techniques. Modern flagship studies are rigorous and multimodal, validating hypotheses by a multitude of distinct methods [31]. It has become increasingly common for such studies to incorporate advanced microscopy, state-of-the-art sequencing technology, and multiple biological models [31]. The resulting datasets continue to increase in size and complexity, necessitating sophisticated and integrative approaches to experimental design and analysis [31]. scRNA-seq contributes to lineage tracing by providing transcriptomic profiles that reveal cellular states and transitions along differentiation trajectories. Computational methods for trajectory inference, such as pseudotime analysis, can reconstruct the sequence of gene expression changes as cells progress along developmental pathways [28]. While these analysis pipelines are rapidly maturing, gold standards for data integration methods and trajectory inference are still lacking, representing an active area of methodological development [28]. The combination of experimental lineage tracing with computational trajectory inference from scRNA-seq data enables robust reconstruction of developmental hierarchies and validation of lineage relationships.
Integrated Experimental Workflows
Comprehensive scRNA-seq Experimental Pipeline
A complete scRNA-seq workflow encompasses multiple stages from experimental design to data interpretation. The process begins with project planning, which requires two principal elements: sequencing data that can be assigned to gene models with functional annotations and putative orthologies, and a protocol for cell or nuclei suspensions from the tissue of interest [28]. The decision to sequence single cells or single nuclei depends on the intended application, with entire cell capture being ideal for capturing cytoplasmic mRNAs, while nuclear isolation benefits difficult-to-isolate cells like neurons and is compatible with multiome studies combining transcriptomes with open chromatin (ATAC-seq) [28]. The experimental phase involves generating single-cell or nuclei suspensions, optimizing dissociation protocols to maintain cell viability while minimizing stress responses, followed by cell capture, library preparation, and sequencing [28]. The computational phase includes quality control, read alignment, count matrix generation, normalization, dimensionality reduction, clustering, and biological interpretation [28]. Throughout this workflow, careful consideration of costs is essential, including sample preparation (tissue dissociation, cell sorting, viability assays), library preparation (reagents and kits for chosen platform), and sequencing costs (driven by read depth per number of cells) [28].
Visualization and Data Analysis Frameworks
The analysis of scRNA-seq data presents unique visualization challenges due to the high-dimensional nature of the data. Traditional visualization methods like t-SNE and UMAP, while widely adopted, face limitations in accurately rendering the geometric relationship of populations with distinct functional states [30]. These methods are not optimal for preserving global geometric structure and may result in contradictions where clusters with near distance in embedded dimensions are further apart in original dimensions [30]. Additionally, UMAP and t-SNE cannot track the variance of clusters effectively, as variance representation becomes proportional to sample size rather than true variance [30]. To address these challenges, supervised visualization methods like supCPM (supervised Capacity Preserved Mapping) have been developed, which separate different clusters, preserve global structure, and track cluster variance [30]. This approach incorporates label information from clustering results to assist visualization, addressing the crowding issue that plagues many classical geometry-preserving methods [30]. The development of such advanced visualization frameworks is particularly important for interpreting complex lineage trajectories and rare cell populations identified through scRNA-seq analyses.
Diagram 1: Integrated scRNA-seq Workflow for Rare Cell Discovery and Lineage Tracing. This diagram illustrates the comprehensive experimental and computational pipeline for scRNA-seq studies, highlighting the parallel paths for rare cell identification and lineage trajectory reconstruction, and their integration into key biological applications.
Research Reagent Solutions
The successful implementation of scRNA-seq and lineage tracing experiments relies on a suite of specialized reagents and tools. The selection of appropriate reagents depends on the specific research goals, sample characteristics, and experimental design. Commercial platforms offer diverse solutions with different strengths and limitations, enabling researchers to tailor their approach to specific project requirements. Similarly, the choice of lineage tracing technologies depends on the desired resolution, specificity, and experimental model. The following table summarizes key research reagent solutions essential for implementing the applications described in this protocol.
Table 3: Essential Research Reagents and Platforms for scRNA-seq and Lineage Tracing
| Category | Specific Products/Systems | Key Features | Applications |
|---|---|---|---|
| scRNA-seq Platforms | 10x Genomics (GEM-X v4) | Droplet microfluidics; 500-20,000 cells per run | Standardized high-throughput scRNA-seq |
| Illumina (Fluent BioSciences) | Vortex-based droplet capture; no size restrictions | Flexible input requirements | |
| BD Rhapsody | Microwell-based capture; larger cell size capacity | Handling delicate or large cells | |
| Parse BioScience | Plate-based combinatorial barcoding; >100,000 cells | Low cost per cell; large scale projects | |
| Lineage Tracing Systems | Cre-loxP | Site-specific recombination; inducible variants | Broad applicability; temporal control |
| Dre-rox | Heterospecific recombinase; compatible with Cre | Dual recombinase systems; complex fate mapping | |
| R26R-Confetti | Multicolour reporter; stochastic expression | Clonal analysis; single-cell resolution | |
| Brainbow | Multicolour reporter cassette; multiple FPs | Visualizing cellular neighborhoods | |
| Analysis Tools | Seurat | R-based toolkit; comprehensive analysis | End-to-end scRNA-seq analysis |
| Scanpy | Python-based toolkit; scalable processing | Large dataset analysis; integration with ML | |
| scMSCF | Multi-scale clustering; transformer model | Enhanced rare cell identification | |
| supCPM | Supervised visualization; preserves structure | Accurate trajectory visualization |
Executing scRNA-seq: A Step-by-Step Guide to Isolation, Library Prep, and Platform Selection
Single-cell isolation represents a foundational step in single-cell RNA sequencing (scRNA-seq) protocols, enabling the resolution of cellular heterogeneity that is often obscured in bulk population analyses [32] [2]. The selection of an appropriate isolation strategy directly impacts key experimental outcomes, including cell viability, recovery efficiency, transcriptomic coverage, and overall data quality [32] [1]. Within the context of developing robust scRNA-seq protocols for isolates research, scientists must navigate a technological landscape comprising several established platforms: fluorescence-activated cell sorting (FACS), microfluidic systems, and droplet-based partitioning [32] [33] [34]. Each methodology offers distinct advantages and limitations regarding throughput, multiplexing capability, cost, and technical accessibility [32] [35]. This application note provides a structured comparison of these single-cell isolation strategies, detailing their underlying principles, standardized protocols, and performance metrics to guide researchers in selecting optimal approaches for their specific experimental requirements in drug development and basic research.
Comparative Analysis of Single-Cell Isolation Platforms
The following table summarizes the key technical specifications and performance characteristics of the three major single-cell isolation platforms.
Table 1: Technical Comparison of Major Single-Cell Isolation Platforms
| Parameter | FACS | Microfluidic Systems | Droplet-Based Systems |
|---|---|---|---|
| Throughput | High (up to thousands of cells) [32] | Medium to High [33] | Very High (thousands to millions of cells) [34] [36] |
| Principle | Electrostatic deflection based on light scattering and fluorescence [32] | Physical manipulation in microfabricated channels/chambers [33] | Microfluidic partitioning into water-in-oil emulsions [34] [36] |
| Multiplexing Capability | High (up to 18 parameters with modern systems) [32] | Limited by design [33] | High, via barcoding strategies [37] [36] |
| Cell Viability | Variable (can be impacted by shear stress) [32] | Generally high [33] | Generally high [36] |
| Typical Cell Capture Efficiency | Requires high starting cell count (>10,000) [32] [33] | Varies by system | 30-75% [36] |
| mRNA Capture Efficiency | N/A (downstream method dependent) | N/A (downstream method dependent) | 10-50% [36] |
| Key Advantages | High specificity, multi-parameter analysis, index sorting [32] [38] | Low reagent consumption, integrated workflows [32] [33] | Unmatched throughput, cost-effectiveness at scale, commercial standardization [34] [36] |
| Key Limitations | High equipment cost, requires specialized expertise, high input requirement [32] [33] | Can require high skill, often dissociated cells only [32] | Multiplet rates (<5%), barcode collisions, ambient RNA contamination [37] [36] |
Detailed Experimental Protocols
Protocol 1: Fluorescence-Activated Cell Sorting (FACS) for scRNA-seq
Principle: FACS utilizes laser-based interrogation of individual cells in a fluid stream. Cells are labeled with fluorescent probes (e.g., antibodies, viability dyes) and electrically charged based on predefined parameters. Charged droplets are then deflected into collection plates for downstream processing [32] [38].
Table 2: Key Reagent Solutions for FACS Protocol
| Reagent/Material | Function |
|---|---|
| Fluorophore-conjugated Antibodies | Target specific surface proteins for cell type selection [32]. |
| Viability Dye (e.g., Propidium Iodide) | Distinguish and exclude non-viable cells [32]. |
| Collection Plate (96-well or 384-well) | Contains lysis buffer to preserve RNA upon cell capture [38]. |
| Sorting Buffer (e.g., PBS with BSA) | Maintains cell viability and stability during the sorting process. |
Step-by-Step Workflow:
- Sample Preparation: Generate a single-cell suspension from your tissue or cell culture. The viability should ideally exceed 85% [1] [36]. Enzymatic or mechanical dissociation must be optimized for the specific tissue to minimize the induction of artificial stress responses [1].
- Staining: Incubate the cell suspension with pre-titrated, fluorophore-conjugated antibodies and a viability dye for 20-30 minutes on ice, protected from light.
- Setup and Calibration: Configure the FACS sorter according to manufacturer guidelines. Create a gating strategy to identify the target population based on size (FSC), granularity (SSC), viability (viability dye negative), and positive expression of fluorescent markers. Include a "single-cell" gate to maximize the recovery of true single cells.
- Index Sorting: Enable the index sorting feature if available. This records the fluorescence parameters of each individually sorted cell, allowing correlation of surface marker expression with downstream transcriptomic data [38].
- Collection: Sort single cells directly into the wells of a 96-well or 384-well plate pre-loaded with an appropriate lysis buffer and RNase inhibitors. Plates should be kept on a cooling block during sorting. The lysis buffer may contain chaotropic agents like Trifluoroethanol (TFE) for efficient lysis and protein denaturation [38].
- Downstream Processing: Immediately proceed to reverse transcription or freeze the plate at -80°C. Standard scRNA-seq library preparation protocols, such as SMART-Seq2, can then be applied [2] [1].
Protocol 2: Droplet-Based Microfluidics (e.g., 10x Genomics)
Principle: This approach uses microfluidic chips to co-encapsulate single cells with barcoded gel beads in nanoliter-scale droplets (GEMs). Within each droplet, cell lysis occurs, and mRNA transcripts are barcoded with a unique cellular identifier and a unique molecular identifier (UMI) during reverse transcription [34] [36].
Table 3: Key Reagent Solutions for Droplet-Based Protocol
| Reagent/Material | Function |
|---|---|
| Barcoded Gel Beads | Source of oligonucleotides with cell barcode, UMI, and poly(dT) for mRNA capture [36]. |
| Partitioning Oil & Reagents | Forms stable water-in-oil emulsions for nanoreactor droplets. |
| Master Mix | Contains reagents for reverse transcription and cell lysis. |
| Single-Cell 3' Reagent Kit (Commercial) | Provides all necessary enzymes and buffers for cDNA synthesis and library construction. |
Step-by-Step Workflow:
- Sample Preparation: Prepare a high-quality single-cell suspension at a recommended concentration of 700-1,200 cells/µL with high viability (>85%) [36]. Accurately determining cell concentration is critical to minimize doublet formation.
- Loading: Load the single-cell suspension, barcoded gel beads, and partitioning oil/master mix into the designated reservoirs of a microfluidic chip (e.g., 10x Genomics Chromium Chip).
- Partitioning: Run the chip on the controller instrument. The microfluidic circuitry generates gel beads-in-emulsion (GEMs), where ideally each GEM contains a single cell and a single gel bead.
- Barcoding and RT: Collect the GEMs in a tube and incubate on a thermal cycler. During this step, cells are lysed, and the gel beads dissolve, releasing barcoded oligonucleotides. mRNA is captured by poly(dT) primers, and reverse transcription occurs, labeling all cDNA from a single cell with the same cellular barcode and each transcript with a UMI [36].
- Breaking Emulsions: The oil emulsion is broken, and the pooled cDNA is cleaned up using magnetic beads.
- Library Preparation: The barcoded cDNA is amplified via PCR, and a sequencing library is constructed by fragmentation, end-repair, adapter ligation, and sample indexing.
- Quality Control and Sequencing: Validate the library quality (e.g., using Bioanalyzer) and sequence on an appropriate Illumina platform.
Protocol 3: Microfluidics-Free Templated Emulsification (PIP-seq)
Principle: A recent innovation, Particle-templated Instant Partition Sequencing (PIP-seq), eliminates the need for specialized microfluidic hardware. It uses vortexing to create monodispersed emulsions by mixing cells, barcoded hydrogel templates, and lysis reagents in standard laboratory tubes or plates [35].
Step-by-Step Workflow:
- Preparation: Mix a single-cell suspension with barcoded hydrogel templates and proteinase K-containing lysis buffer in a tube or well plate. The proteinase K is inactive at low temperatures.
- Emulsification: Vortex the mixture vigorously for ~2 minutes to generate a uniform water-in-oil emulsion, compartmentalizing single cells with barcoded beads.
- Thermal Activation: Incubate the emulsion at 65°C to activate proteinase K, leading to cell lysis and release of mRNA for capture on the barcoded beads.
- Storage or Processing: Emulsions can be stored at 0°C for days or processed immediately.
- Reverse Transcription: Remove the oil, transfer the beads to a reverse transcription buffer, and synthesize full-length cDNA.
- Library Construction: Amplify the cDNA and prepare libraries for sequencing following protocols similar to other droplet-based methods [35].
Practical Implementation and Guidance
Application-Specific Workflow Selection
Choosing the optimal isolation strategy depends heavily on the research goals and sample constraints. The following diagram outlines a decision-making workflow to guide researchers.
Troubleshooting Common Challenges
- Minimizing Doublets and Multiplets: In droplet-based systems, accurate cell concentration measurement is paramount to avoid overloading, which increases multiplet rates [37]. For FACS, proper calibration of the single-cell gate is essential. Techniques like Cell Hashing or MULTI-seq can be employed to demultiplex and identify doublets computationally [37].
- Reducing Ambient RNA: Ambient RNA from lysed cells can contaminate transcriptome data. Strategies include using viability dyes during sorting to remove dead cells, incorporating enzymatic degradation steps (e.g., RNase A treatment) in droplet protocols, and employing computational tools like SoupX or DecontX to subtract ambient signals post-sequencing [37].
- Improving mRNA Capture Efficiency: The use of unique molecular identifiers (UMIs) is critical to correct for amplification biases and enable accurate transcript counting, thereby improving quantitative accuracy despite imperfect capture [1] [36]. Optimizing lysis buffer composition (e.g., incorporating TFE) can also enhance efficiency [38].
The strategic selection of a single-cell isolation method is a critical determinant of success in scRNA-seq studies. FACS remains the gold standard for presorting specific cell populations based on surface markers, while droplet-based microfluidics offers unparalleled throughput for large-scale atlas projects. Emerging microfluidics-free technologies like PIP-seq promise greater scalability and accessibility. As the field progresses, the integration of these isolation techniques with multi-omic readouts (e.g., CITE-seq, ATAC-seq) and spatial transcriptomics will provide an even more comprehensive view of cellular identity and function, further empowering research and drug development [37] [36]. By aligning the technical capabilities of each platform with specific experimental requirements, researchers can effectively harness the power of single-cell genomics to unravel biological complexity.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical research by allowing scientists to probe the transcriptomes of individual cells, revealing previously unappreciated levels of cellular heterogeneity and complexity [1] [2]. Since its conceptual breakthrough in 2009, the technology has rapidly evolved, enabling the analysis of hundreds of thousands of cells in a single experiment and fueling discoveries across areas such as cancer progression, embryonic development, and regenerative medicine [1]. The core wet-lab workflow of scRNA-seq involves a series of critical steps: the effective isolation of viable single cells or nuclei, cell lysis to release RNA molecules, reverse transcription to convert RNA into complementary DNA (cDNA), and cDNA amplification to generate sufficient material for sequencing [1] [2]. The precision and optimization of these initial steps are paramount, as they directly impact the sensitivity, accuracy, and overall quality of the final sequencing data. This application note provides a detailed protocol for these foundational wet-lab procedures, framed within the context of a robust scRNA-seq pipeline for isolated cells and nuclei.
scRNA-seq Workflow: From Cell to Library
The following diagram illustrates the core pathway from a fresh sample to a sequenced single-cell RNA library, highlighting the key wet-lab steps covered in this protocol.
Detailed Experimental Protocols
Single-Cell or Single-Nucleus Isolation
The first critical step is obtaining a high-quality suspension of single cells or nuclei. The choice between whole cells or nuclei is sample-dependent. Single-nucleus RNA-seq (snRNA-seq) is particularly advantageous for frozen samples, tissues that are difficult to dissociate (e.g., brain), or when aiming to minimize artifactual stress responses induced by tissue dissociation procedures [1].
Protocol for Single-Nucleus Isolation from Frozen PAXgene Blood Specimens (Adapted from CL-D snRNAseq Protocol [39])
- Sample Input: Frozen whole blood collected in PAXgene Blood RNA Tubes.
- Cell Lysis Buffer Preparation: Prepare a lysis buffer according to Table 1. Key components include detergents (NP-40, Tween-20) for membrane disruption, MgCl₂ and Tris-HCl for ionic and pH stability, RNase Inhibitor to preserve RNA integrity, and BSA to stabilize the nuclei.
- Procedure:
- Thaw the frozen PAXgene blood sample on ice.
- Add a pre-cooled cell lysis buffer to the sample and mix thoroughly by pipetting.
- Incubate the mixture on ice for a specified duration (e.g., 5-10 minutes) to complete the lysis of cellular membranes while leaving nuclei intact.
- Filter the lysate through a series of pre-cooled cell strainers (e.g., 100µm followed by 40µm) to remove large debris and aggregates.
- Centrifuge the filtered suspension at 4°C at 500 RCF for 5 minutes to pellet the nuclei.
- Carefully discard the supernatant and resuspend the nuclei pellet in a wash buffer (for formulation, see Table 2).
- Count the nuclei and assess viability using a cell counter (e.g., Cellometer Auto 2000) with appropriate nuclear stains.
Cell Lysis and RNA Release
Once individual cells or nuclei are isolated, the next step is lysis to release RNA. The composition of the lysis buffer is crucial, as it must efficiently release RNA while inactivating RNases and being compatible with downstream enzymatic reactions [39] [40].
Detailed Lysis Protocol for Single Cells [39] [2]
- Principle: The lysis buffer uses a combination of ionic and non-ionic detergents to dissolve lipid membranes and release cellular contents. RNase inhibitors are critical additives to prevent RNA degradation.
- Lysis Buffer Formulation: Table 1 provides a detailed recipe for a robust lysis buffer suitable for single cells or nuclei.
- Procedure:
- Prepare the lysis buffer fresh and keep it on ice.
- Resuspend the purified cell or nucleus pellet in the prepared lysis buffer by gentle pipetting.
- Incubate the suspension on ice for a defined period (typically 5-10 minutes) to ensure complete lysis.
- For single-nucleus protocols, a centrifugation step may follow to pellet nuclei. However, note that centrifugation can deplete specific mRNA classes (e.g., those encoding cytoskeletal proteins) that may be associated with insoluble cellular components [40]. For whole-cell lysis, the lysate can often proceed directly to reverse transcription without a clarification spin.
Table 1: Lysis Buffer Preparation (Volumes in ml) [39]
| Component | Final Volume: 1ml | Final Volume: 2ml (4 reactions) | Final Volume: 4ml |
|---|---|---|---|
| H₂O | 0.835 | 1.67 | 3.34 |
| 5M NaCl | 0.002 | 0.004 | 0.008 |
| 1M Tris-HCl pH7.4 | 0.010 | 0.020 | 0.040 |
| 1M MgCl₂ | 0.003 | 0.006 | 0.012 |
| 10% Tween-20 | 0.010 | 0.020 | 0.040 |
| Freshly prepared 10% NP-40 | 0.010 | 0.020 | 0.040 |
| Digitonin | 0.002 | 0.004 | 0.008 |
| 10% MACS BSA Stock Solution | 0.100 | 0.200 | 0.400 |
| DTT | 0.001 | 0.002 | 0.004 |
| RNase Inhibitor | 0.025 | 0.050 | 0.100 |
Reverse Transcription (RT)
Reverse transcription converts the released RNA, specifically polyadenylated mRNA, into more stable cDNA. This step incorporates essential sequences for downstream sequencing and quantification.
Protocol for Reverse Transcription with Barcoding and UMIs [1] [2] [41]
- Principle: Poly(T) primers anneal to the poly(A) tails of mRNA. These primers also contain:
- Cell Barcodes: A unique DNA sequence that tags all cDNA from a single cell, allowing samples to be pooled for sequencing.
- Unique Molecular Identifiers (UMIs): A random short sequence that labels individual mRNA molecules, enabling accurate digital counting and eliminating PCR amplification bias [1].
- Procedure:
- To the cell lysate, add a master mix containing reverse transcriptase, dNTPs, and the custom barcoded poly(T) primers.
- Incubate the reaction at appropriate temperatures (e.g., 42°C for 60-90 minutes) to synthesize first-strand cDNA.
- Inactivate the reverse transcriptase by heating.
cDNA Amplification
The minute amounts of cDNA generated from a single cell must be amplified to produce a sequencing library.
Protocol for cDNA Amplification via PCR [1] [2]
- Principle: The full-length cDNA is amplified using PCR. A common method leverages the "template-switching" effect of certain reverse transcriptases, which adds a universal adapter sequence to the 3' end of the cDNA, allowing for PCR amplification with a single primer pair.
- Procedure:
- Add a PCR master mix containing a DNA polymerase, primers complementary to the universal adapters on the cDNA, and dNTPs.
- Run a PCR with a cycle number optimized to amplify the cDNA sufficiently without introducing excessive duplication bias (typically 12-18 cycles).
- Purify the amplified cDNA using solid-phase reversible immobilization (SPRI) beads.
- Assess the quality and quantity of the amplified cDNA using a Bioanalyzer or similar instrument.
Table 2: General Wash Buffer (PBS with 0.04% BSA) Preparation (Volumes in ml) [39]
| Final Volume: | 1 ml | 50 ml | 100 ml | 150 ml (4 reactions) |
|---|---|---|---|---|
| PBS | 0.996 | 49.8 | 99.6 | 149.4 |
| 10% BSA (Miltenyi) | 0.004 | 0.2 | 0.4 | 0.6 |
| RNase Inhibitor | 0.001 | 0.05 | 0.1 | 0.15 |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and their critical functions in the scRNA-seq wet-lab workflow.
Table 3: Key Research Reagent Solutions for scRNA-seq
| Reagent / Solution | Function / Application |
|---|---|
| RNase Inhibitor | Prevents degradation of RNA during cell lysis and subsequent steps, crucial for preserving transcriptome integrity [39]. |
| Non-ionic Detergents (e.g., NP-40, Tween-20, Triton X-100) | Solubilize cell and nuclear membranes to release RNA, while maintaining the activity of enzymes used in downstream reactions [39] [40]. |
| Bovine Serum Albumin (BSA) | Stabilizes nuclei and proteins, reduces nonspecific binding in buffers, and improves overall reaction efficiency [39]. |
| Dithiothreitol (DTT) | A reducing agent that helps break disulfide bonds in proteins, contributing to more effective cell lysis [39]. |
| Barcoded Poly(T) Primers with UMIs | Primers for reverse transcription that incorporate a cell-specific barcode and unique molecular identifier on each mRNA molecule, enabling sample multiplexing and accurate transcript quantification [1] [41]. |
| Template Switching Oligo (TSO) | Used in conjunction with specific reverse transcriptases to add a universal primer sequence to the 5' end of cDNA, facilitating full-length cDNA amplification [42]. |
The wet-lab journey from a complex biological sample to a sequenced single-cell library is a technically demanding but rewarding process. Mastery of the core steps—effective cell or nucleus isolation, optimized lysis, efficient reverse transcription, and faithful cDNA amplification—forms the bedrock of any successful scRNA-seq study. The protocols and reagents detailed here provide a reliable foundation. However, researchers must remember that these protocols often require sample-specific optimization, particularly for challenging tissues or unique experimental conditions. As the field continues to advance, these foundational wet-lab techniques will remain essential for unlocking the profound insights into cellular heterogeneity and function that single-cell genomics promises.
Single-cell RNA sequencing (scRNA-seq) represents a transformative methodology that enables the investigation of transcriptional profiles at the ultimate level of biological resolution: the individual cell. This technology has moved beyond bulk RNA-seq, which averages gene expression across thousands to millions of cells, to reveal the cellular heterogeneity, rare cell populations, and dynamic transitions that underlie complex biological systems [1] [2]. A fundamental distinction in scRNA-seq experimental design lies in choosing between high-throughput and low-throughput platforms, a decision that profoundly impacts the scale, depth, and ultimate conclusions of a study.
The selection between these approaches is not merely a technical preference but must be strategically aligned with the core research question. High-throughput methods are engineered to profile hundreds to millions of cells, providing unparalleled breadth in capturing cellular diversity within complex tissues [43] [12]. Conversely, low-throughput methods focus on dozens to hundreds of cells, typically delivering greater sequencing depth and more complete transcriptomic information per cell, which is crucial for detecting subtle transcriptional differences, splice variants, and low-abundance transcripts [2] [44]. This application note provides a structured comparison and detailed experimental protocols to guide researchers in matching the appropriate scRNA-seq platform to their specific research needs within the context of studying isolated cells.
Platform Comparison: Technical Specifications and Performance
The performance characteristics of scRNA-seq platforms vary significantly, influencing their suitability for different research scenarios. The following table summarizes key metrics and optimal use cases for the main platform types.
Table 1: Comparison of scRNA-seq Platform Types
| Feature | High-Throughput (Droplet/Microwell-Based) | Low-Throughput (Plate/Fluidics-Based) |
|---|---|---|
| Typical Cell Throughput | Hundreds to tens of thousands of cells per run [43] [2] | Dozens to hundreds of cells per run [2] |
| Sequencing Depth | Lower depth per cell (typically 3' or 5' focused) [45] | Higher depth per cell; often full-length transcript coverage [45] [46] |
| Key Applications | Identifying cellular heterogeneity, rare cell types, building atlases [47] [2] | Studying splice variants, sequence mutations, in-depth analysis of few cells [2] [44] |
| Cell Capture Method | Microfluidic droplets (e.g., 10x Genomics, MobiDrop) or microwells [48] [12] | Microfluidic traps (e.g., Fluidigm C1), FACS, or manual plating [45] |
| Representative Platforms | 10x Genomics Chromium X, MobiNova-100, SeekOne, BGI C4 [48] | Fluidigm C1, WaferGen iCell8 [45] |
| Cost per Cell | Lower | Higher |
| Protocol Flexibility | Standardized, fixed protocols | More adaptable for customization |
Recent comparative studies have quantified the performance of specific high-throughput platforms. For instance, a 2025 benchmark study comparing droplet-based systems on Peripheral Blood Mononuclear Cells (PBMCs) reported that the 10x Genomics Chromium X platform captured a median of 2,085 genes and 5,532 UMIs per cell, while the MobiNova-100 platform showed competitive performance, closely following Chromium X in comprehensive assessments [48]. Such empirical data is critical for informing platform selection based on required sensitivity.
Table 2: Performance Metrics of Selected Commercial High-Throughput Platforms (PBMC Test)
| Platform | Median Genes per Cell | Median UMIs per Cell | Notable Strength |
|---|---|---|---|
| 10x Genomics Chromium X | 2,085 | 5,532 | Excellent overall performance [48] |
| MobiNova-100 | 1,747 | 4,365 | Significance in differential gene expression [48] |
| SeekOne Platform | 1,782 | 5,172 | Close UMI performance to market leader [48] |
Experimental Protocols
Core Experimental Workflow
The journey from a biological sample to single-cell data follows a multi-stage pathway. The initial steps diverge based on the chosen platform technology, but later stages converge for sequencing and analysis. The diagram below outlines this core workflow, highlighting the key decision points and procedures.
Protocol A: High-Throughput Droplet-Based scRNA-seq (e.g., 10x Genomics Chromium X, MobiNova-100)
This protocol is designed for capturing transcriptomes from thousands of cells in a single run, ideal for comprehensive profiling of heterogeneous samples [48] [12].
1. Sample Preparation: Generating High-Quality Single-Cell Suspension
- Tissue Dissociation: Use a combination of enzymatic and mechanical dissociation appropriate for the specific tissue. For fragile tissues, consider cold-active proteases and perform dissociation at 4°C to minimize artifactual stress responses [1] [44].
- Cell Staining and Viability Assessment: Stain cells with a viability dye (e.g., Calcein AM, Propidium Iodide). Assess viability and count using a hemocytometer or automated cell counter. Critical: Target >80% viability for optimal droplet encapsulation [45].
- Cell Suspension Adjustment: Centrifuge and resuspend cells in an appropriate buffer (e.g., PBS with 0.04% BSA). Filter the suspension through a flow cytometry-compatible strainer (30-40 µm) to remove cell clumps and debris. Adjust concentration to the target recommended by the platform (e.g., 700–1,200 cells/µL for Chromium X) [48] [12].
2. Library Preparation: Droplet Partitioning and Barcoding
- Gel Bead-in-Emulsion (GEM) Generation: Load the single-cell suspension, barcoded Gel Beads, and partitioning oil into a microfluidic chip. The Chromium X or MobiNova-100 instrument will combine these components to generate nanoliter-scale droplets, each theoretically containing a single cell and a single gel bead [48] [12].
- Within-Droplet Reactions: Inside each droplet:
- Cell Lysis: The cell membrane is lysed, releasing RNA.
- Reverse Transcription (RT): The gel bead dissolves, releasing oligos with poly(dT) primers, cell barcodes, and Unique Molecular Identifiers (UMIs). Reverse transcription occurs, creating barcoded cDNA from polyadenylated mRNA [12].
- cDNA Recovery and Amplification: Break the droplets and pool the contents. Purify the barcoded cDNA using magnetic beads. Amplify the cDNA via PCR to generate sufficient material for library construction [48].
- Library Construction and Sequencing: Fragment the amplified cDNA, add sequencing adaptors, and index via a second PCR. Purify the final library and validate quality using a Bioanalyzer or TapeStation. Quantify using fluorometry (e.g., Qubit). Pool libraries and sequence on an Illumina platform (e.g., NovaSeq) with a read configuration suitable for the platform (e.g., 28 bp Read 1 for barcodes/UMI, 91 bp Read 2 for transcript) [48] [45].
Protocol B: Low-Throughput Plate-Based scRNA-seq (e.g., Fluidigm C1, SMART-seq)
This protocol prioritizes transcriptome depth and completeness from a limited number of cells, suitable for focused studies on pre-defined cell populations [2] [44].
1. Single-Cell Isolation and Capture
- Cell Sorting via FACS: Prepare a single-cell suspension as in Protocol A. Use Fluorescence-Activated Cell Sorting (FACS) to sort individual, viable cells directly into the wells of a 96- or 384-well plate containing lysis buffer. Precise gating based on size, granularity, and viability dye is crucial. Include a control well without a cell for contamination monitoring [47] [45].
- Alternative Capture (Fluidigm C1): Load the cell suspension onto a primed, integrated fluidic circuit (IFC) chip designed for a specific cell size range. The C1 system will automatically trap individual cells in microfluidic chambers via fluidic resistance. Image the chip to confirm single-cell occupancy in each capture site [45].
2. On-Plate/On-Chip Reverse Transcription and cDNA Amplification
- Cell Lysis and Poly(A) RNA Capture: Lyse cells in the plate/IFC chip. The lysis buffer should contain detergents and RNase inhibitors.
- Reverse Transcription and cDNA Amplification: Use a protocol that allows for full-length transcript coverage, such as SMART-seq2 [46].
- The reverse transcriptase adds a few non-templated nucleotides to the 3' end of the cDNA.
- A template-switching oligo (TSO) binds to these nucleotides, allowing the reverse transcriptase to continue replication, thereby adding a universal adapter sequence to the end of the cDNA.
- The cDNA is then amplified by PCR using primers targeting the universal adapter. This method efficiently amplifies full-length transcripts [1] [44].
3. Library Preparation and Sequencing
- Library Construction: Tagment the amplified cDNA using a transposase-based kit (e.g., Nextera XT) to fragment the DNA and add sequencing adapters in a single step. Alternatively, use mechanical fragmentation followed by standard library preparation.
- Quality Control and Sequencing: Pool the individually indexed libraries. Validate the pooled library's size distribution and quantity. Sequence on an Illumina platform to a sufficient depth (typically higher than high-throughput methods) to leverage the full-length information, using a paired-end read configuration (e.g., 2x150 bp) [45].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful scRNA-seq experiments rely on a suite of specialized reagents and kits. The table below details key components and their functions.
Table 3: Essential Reagents and Kits for scRNA-seq Workflows
| Reagent/Kit Name | Function | Example Use Case |
|---|---|---|
| Single Cell 3' or 5' Kit (e.g., 10x Genomics, MobiDrop) | Integrated reagent kit for droplet-based platforms; contains chips, gel beads, buffers for GEM generation, RT, and library prep. | High-throughput profiling of PBMCs or tumor dissociates for atlas building [48] [12]. |
| SMARTer Ultra Low RNA Kit | Technology for cDNA synthesis and amplification from single cells, utilizing template-switching for full-length coverage. | Low-throughput, plate-based studies requiring full-length transcript data (e.g., SMART-seq2) [45]. |
| Nextera XT DNA Library Preparation Kit | Transposase-based kit for simultaneous fragmentation and adapter tagging of cDNA libraries. | Preparing sequencing libraries from amplified cDNA in low-throughput workflows [45]. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences that uniquely tag each mRNA molecule during RT, enabling accurate quantification and removal of PCR duplicates. | Essential for quantitative analysis in both high- and low-throughput methods [1]. |
| Viability Staining Dyes (e.g., Calcein AM, Propidium Iodide) | Fluorescent dyes to distinguish live (metabolically active) cells from dead cells prior to capture. | Critical quality control step before loading cells onto any platform to ensure data quality [45]. |
Decision Framework: Matching Platform to Biological Question
The choice between high-throughput and low-throughput methodologies should be a deliberate one, guided by the primary objective of the study. The following diagram provides a logical pathway for making this critical decision.
Guidelines for Platform Selection
- Choose a High-Throughput Platform When: The research aims to comprehensively catalog cell types and states within a complex tissue, discover rare populations (e.g., cancer stem cells, rare immune subsets), or construct a cellular atlas. This is the default for exploratory studies of heterogeneous samples where the full spectrum of diversity is unknown [2] [12].
- Choose a Low-Throughput Platform When: The research focuses on a small, well-defined set of cells where deep molecular characterization is required. This includes studies of alternative splicing, allele-specific expression, viral RNAs, or the detection of low-abundance transcripts. It is also suitable when cells are extremely limited but each is of high value [2] [44].
- Consider a Hybrid or Combined Approach: For large-scale projects, an effective strategy is to use a high-throughput platform to map the overall heterogeneity and identify key populations of interest, followed by a low-throughput, deep-sequencing approach on sorted populations from these groups for in-depth molecular analysis [46].
The dichotomy between high-throughput and low-throughput scRNA-seq platforms represents a strategic choice between breadth and depth. High-throughput droplet-based methods provide a wide-angle lens to survey cellular ecosystems, while low-throughput plate-based methods offer a powerful zoom to scrutinize specific cellular features. There is no universally superior platform; the optimal instrument is the one that is precisely matched to the biological question at hand. By applying the decision framework and protocols outlined in this document, researchers can design more robust and informative single-cell studies, thereby maximizing the return on investment and accelerating discovery in biomedical research and drug development.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical research by enabling the characterization of gene expression at the fundamental unit of biology—the individual cell. This capability reveals cellular heterogeneity, identifies rare cell populations, and uncovers novel biological insights that are obscured in bulk RNA-seq analysis of tissue ensembles [2]. The field has diversified into several methodological approaches, each with distinct strengths, limitations, and optimal applications. This application note provides a detailed technical comparison of three foundational scRNA-seq strategies: SMARTer-based full-length protocols, droplet-based methods (exemplified by 10x Genomics), and combinatorial indexing techniques. Understanding these methods' core principles and technical nuances is essential for selecting the appropriate protocol for specific research goals in drug development and basic science.
The following table summarizes the key characteristics, advantages, and limitations of the three scRNA-seq methods discussed in this note.
Table 1: Technical Comparison of scRNA-seq Methods
| Feature | SMARTer-based (e.g., Smart-seq3xpress) | Droplet-based (e.g., 10x Genomics) | Combinatorial Indexing (e.g., sci-RNA-seq3) |
|---|---|---|---|
| Core Principle | Plate-based; full-length transcript coverage in nanoliter volumes [49] | Microfluidic partitioning of single cells with barcoded beads in droplets [12] | Combinatorial barcoding of transcripts in permeabilized cells/nuclei across multiple rounds [50] |
| Throughput | Throughput limited by PCR instrumentation [49] | High (80K–960K cells per kit with Chromium X) [12] | Ultra-high (e.g., ~380,000 nuclei in a single experiment) [50] |
| Transcript Coverage | Full-length; enables isoform and SNP analysis [49] [51] | 3' or 5' end counting only [49] [19] | Typically 3' end counting [50] |
| Sensitivity | High genes/cell; detects low-abundance transcripts [49] [51] | Lower genes/cell compared to full-length methods [51] | Varies; optimized protocols show improved sensitivity [50] |
| Key Advantage | Full-transcript coverage and high sensitivity [49] | High cellular throughput and ease of use [12] | Extreme scalability and very low cost per cell at scale [50] |
| Primary Limitation | Lower cellular throughput [49] | Limited transcript information (only ends) [49] | Complex protocol with potential for barcode collisions [50] |
| Cost Efficiency | Reduced reagent use from miniaturization [49] | Commercial kit-based costs | Very low cost per cell (on the order of 1 cent or less) [50] |
Detailed Experimental Protocols
SMARTer-based Method: Smart-seq3xpress
Principle: This protocol miniaturizes the Smart-seq3 workflow to nanoliter volumes, enabling full-length transcript coverage with improved throughput and reduced reagent costs [49].
Workflow:
- Cell Lysis and Reverse Transcription: Single cells are sorted via FACS into 300 nL of lysis buffer covered with an inert hydrophobic substance (e.g., Vapor-Lock) to prevent evaporation [49].
- Reverse Transcription (RT): The RT reaction is performed in a total volume of 400 nL (300 nL lysis + 100 nL RT reagents). The protocol utilizes a template-switching oligonucleotide (TSO) to ensure full-length cDNA synthesis. An improved TSO sequence is employed to reduce strand invasion artifacts [49].
- cDNA Amplification: The pre-amplification PCR is conducted directly by adding 600 nL of PCR mix, using SeqAmp polymerase for optimal compatibility with subsequent tagmentation. The number of PCR cycles can be reduced (e.g., 12 cycles) without compromising gene detection [49].
- Library Construction (Tagmentation): The amplified cDNA is tagmented directly without a clean-up step. The protocol is robust to large variations in cDNA input, allowing for substantial savings in Tn5 transposase usage [49].
- Library Pooling and Sequencing: Final libraries are pooled, typically using a 3D-printed adapter for centrifugation, and are ready for sequencing. The entire process from cell sorting to sequencing-ready libraries can be completed in a single workday [49].
Droplet-based Method: 10x Genomics Chromium
Principle: This high-throughput method uses microfluidics to co-encapsulate single cells with barcoded gel beads and reagents within nanoliter-scale droplets, enabling parallel processing of thousands of cells [12].
Workflow:
- Single-Cell Suspension: Prepare a high-viability single-cell suspension. Cell quality is critical for data quality [12].
- GEM Generation: On a microfluidic chip (e.g., Chromium X), single cells, Gel Beads with barcoded oligonucleotides, and RT reagents are combined with oil to form Gel Beads-in-emulsion (GEMs). The GEM-X technology generates twice as many GEMs at smaller volumes, reducing multiplet rates [12].
- Within-GEM Reactions: Each cell is lysed within its GEM. The Gel Bead dissolves, releasing barcoded oligos that prime the polyadenylated RNA. Reverse transcription produces barcoded cDNA, where all cDNA from a single cell shares the same barcode [12].
- Library Prep: GEMs are broken, and the barcoded cDNA is pooled. The library is constructed via enzymatic fragmentation, sample index PCR, and purification. The process is highly automated and kit-based [12].
- Sequencing and Analysis: Libraries are sequenced on NGS platforms. Data is processed through the Cell Ranger pipeline to generate a gene-cell expression matrix, which can be visualized and explored with tools like Loupe Browser [12].
Combinatorial Indexing Method: sci-RNA-seq3
Principle: This method uses multiple rounds of combinatorial barcoding in a split-pool format to uniquely label the transcripts from hundreds of thousands of fixed cells or nuclei, without the need for physical isolation of single cells [50].
Workflow (Optimized Protocol):
- Nuclei Isolation and Fixation: Nuclei are isolated and fixed with DSP/methanol, which improves nuclear integrity and access to transcripts compared to paraformaldehyde [50].
- RNase Inactivation: A critical step for RNase-rich tissues (e.g., adult). Lysis is performed in a hypotonic phosphate buffer with Diethyl Pyrocarbonate (DEPC), which is more effective than commercial RNase inhibitors for these samples [50].
- First Indexing (Round 1): Fixed nuclei are distributed into a 96-well plate. Reverse transcription is performed using well-specific barcoded oligo-dT primers [50].
- Pool and Split (Round 2): Nuclei from all wells are pooled and randomly split into a new 96-well plate. A second barcode is ligated to the cDNA. The USER enzyme step from the original protocol has been eliminated [50].
- Pool and Split (Round 3): Nuclei are pooled and split again into a third plate. Second-strand synthesis is performed. The nuclei are then digested with a heat-inactivatable protease to extract the cDNA, replacing a costly SPRI bead clean-up [50].
- Tagmentation and Amplification: The extracted cDNA is tagmented with Tn5 transposase, and PCR amplification adds the final index. The final library is purified and sequenced [50].
Visual Workflow Diagrams
The following diagrams illustrate the core procedural and logical workflows for each method.
Diagram 1: The Smart-seq3xpress workflow involves sorting single cells into nanoliter wells, followed by sequential reactions under an oil overlay to minimize volumes and reagent use, culminating in direct tagmentation for library preparation [49].
Diagram 2: The 10x Genomics workflow co-encapsulates single cells with uniquely barcoded beads in droplets (GEMs) for parallel cell lysis and barcoding, followed by bulk library preparation of the pooled cDNA [12].
Diagram 3: The sci-RNA-seq3 workflow uses a split-pool approach where fixed nuclei are subjected to multiple rounds of barcoding. Each nucleus receives a unique combination of barcodes (Barcode 1 + Barcode 2 + Index 3), allowing computational demultiplexing of hundreds of thousands of nuclei from a pooled library [50].
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of scRNA-seq protocols requires careful selection of key reagents. The table below details critical components, their functions, and technology-specific considerations.
Table 2: Key Research Reagent Solutions for scRNA-seq
| Reagent / Material | Core Function | Protocol-Specific Notes |
|---|---|---|
| Template Switching Oligo (TSO) | Enables full-length cDNA synthesis during reverse transcription by adding a universal primer binding site. | Smart-seq3: Critical; improved TSO designs reduce mis-priming artifacts [49]. |
| Tn5 Transposase | An engineered transposase that simultaneously fragments and tags double-stranded DNA with adapters. | Smart-seq3xpress & sci-RNA-seq3: Used for tagmentation; amounts can be optimized for cost reduction [49] [50]. |
| Barcoded Beads | Microbeads conjugated to oligonucleotides containing cell barcode, UMI, and PCR adapter. | 10x Genomics: Core to the technology; each bead has a unique barcode sequence [12]. |
| Polymerase | Enzymatic amplification of cDNA. | Smart-seq3xpress: SeqAmp polymerase is preferred for direct tagmentation compatibility [49]. sci-RNA-seq3: Standard polymerases are used in the PCR amplification step [50]. |
| RNase Inhibitor | Protects RNA molecules from degradation during sample preparation. | sci-RNA-seq3: DEPC is highly effective for inactivating RNases in challenging tissues, replacing expensive commercial inhibitors [50]. |
| Fixative | Preserves cellular state and enables sample storage. | sci-RNA-seq3: DSP/methanol fixation improves nuclear integrity and UMI recovery compared to PFA [50]. |
The choice between SMARTer-based, droplet-based, and combinatorial indexing scRNA-seq methods is not a matter of identifying the "best" technology, but rather of selecting the most appropriate tool for a specific biological question and experimental context. Researchers must weigh the trade-offs between transcriptome depth, cellular throughput, and cost.
- SMARTer-based methods (Smart-seq3xpress) are the gold standard for applications requiring the deepest molecular characterization per cell, such as isoform usage, somatic mutation detection, and detailed gene regulatory network inference [49] [51].
- Droplet-based methods (10x Genomics) offer a robust, user-friendly platform for high-throughput cell atlas construction, immune profiling, and large-scale screening studies where capturing population heterogeneity is the primary goal [12].
- Combinatorial indexing methods (sci-RNA-seq3) provide unparalleled scalability and cost-efficiency for projects requiring the profiling of millions of cells or nuclei, such as whole-organism atlases, and are uniquely suited for frozen or difficult-to-dissociate archival samples [50].
As the field progresses, emerging technologies that combine elements of these approaches—such as FIPRESCI, which integrates combinatorial indexing with droplet microfluidics for massive-scale 5'-end sequencing—are pushing the boundaries of scale and multimodality even further [19]. By understanding the detailed protocols and fundamental principles outlined in this application note, researchers and drug developers can make informed decisions to optimally leverage single-cell genomics in their pursuit of scientific and therapeutic breakthroughs.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical research by enabling the exploration of gene expression at unprecedented resolution, revealing cellular heterogeneity, identifying rare cell populations, and illuminating complex biological systems [21] [1]. However, the transformative potential of scRNA-seq is critically dependent on sample quality and the appropriateness of processing methodologies. The fundamental challenge stems from the inherent fragility of RNA transcripts and the vast differences in tissue composition across biological specimens [52]. Different sample types—fresh, frozen, or challenging-to-dissociate tissues—each present unique technical hurdles that must be systematically addressed to ensure reliable data generation.
The choice of processing method directly impacts key quality metrics in scRNA-seq data, including cell viability, transcriptome complexity, mitochondrial read percentage, and the accurate representation of cellular diversity [53] [52]. For instance, improper handling of fresh tissues can induce artificial stress responses that obscure true biological signals, while suboptimal processing of frozen specimens may yield insufficient RNA quality for meaningful analysis [1] [52]. This protocol provides a comprehensive framework for tailoring scRNA-seq approaches based on sample specifics, enabling researchers to navigate the complex landscape of tissue processing while maximizing data quality and biological insights across diverse experimental contexts.
Sample Types and Their Characteristics
Understanding the inherent properties of different sample types is essential for selecting appropriate processing methodologies in scRNA-seq workflows. Each sample category presents distinct advantages and limitations that directly influence experimental outcomes.
Table 1: Comparison of scRNA-Seq Sample Types and Their Characteristics
| Sample Type | Key Advantages | Major Limitations | Optimal Applications |
|---|---|---|---|
| Fresh Tissues | Preserves full transcriptome including cytoplasmic RNAs; high RNA integrity [1] [52] | Requires immediate processing; dissociation-induced stress responses; logistical challenges [1] [52] | Immunophenotyping; cellular interaction studies; when maximum gene detection is critical |
| Frozen Tissues | Flexibility in processing timing; preserves tissue archives; enables batch experiments [53] [52] | Potential ice crystal damage; reduced RNA quality; lower cytoplasmic RNA content [53] | Large-scale cohort studies; retrospective analysis; biobank utilization |
| FFPE Tissues | Excellent morphological preservation; vast archival resources; room temperature storage [53] | RNA fragmentation and cross-linking; sequence artifacts; biased 3' coverage [53] | Clinical retrospective studies; pathology correlation; when morphology is paramount |
| Challenging Tissues | Access to difficult-to-study tissues; minimal dissociation artifacts [1] [52] | Lower mRNA content; nuclear transcriptome only; may miss key biological processes [1] | Neuronal tissues; fatty tissues; fibrotic tissues; hard-to-dissociate samples |
The quantitative impact of storage conditions on RNA quality is substantial. A systematic comparison of paired fresh, frozen, and FFPE tissues demonstrated that while global gene expression patterns remain correlated (ρ > 0.94 for protein-coding transcripts), significant differences emerge in specific RNA categories [53]. FFPE samples yield highly degraded RNA with increased proportions of intronic and intergenic reads, while mitochondrial and long non-coding RNAs show particular sensitivity to storage conditions [53]. These technical variations must be considered when designing experiments and interpreting resulting data.
Methodologies and Experimental Protocols
Processing Workflow for Fresh Tissues
Success with fresh tissues requires optimized dissociation protocols tailored to specific tissue characteristics and rapid processing to maintain RNA integrity.
Table 2: Customized Dissociation Protocols for Fresh Tumors [52]
| Tumor Type | Recommended Enzymatic Mixture | Key Tissue Characteristics | Recovered Cell Types |
|---|---|---|---|
| Non-Small Cell Lung Cancer (NSCLC) | Liberase TM + Elastase (LE) OR Pronase+Dispase+Elastase+Collagenases A/4 (PDEC) [52] | Complex extracellular matrix; diverse cellular composition [52] | Malignant, immune, fibroblasts, endothelial [52] |
| Glioblastoma (GBM) | Papain (cysteine protease) + DNase I [52] | Delicate neural tissue; sensitive cells [52] | Malignant, neural, immune cells [52] |
| Metastatic Breast Cancer | Liberase TM + DNase I [52] | Collagen-rich matrix; dense tissue [52] | Malignant, immune, stromal cells [52] |
| General Protocol | Enzymes selected based on ECM composition + DNase I to reduce viscosity [52] | Varies by tissue origin and pathology [52] | Cell diversity depends on protocol efficacy [52] |
The dissociation process must balance sufficient tissue disruption with preservation of cell viability and RNA integrity. Different enzymatic approaches recover markedly different cellular diversity even from the same tumor specimen. For example, in NSCLC, only the PDEC and LE protocols successfully recovered fibroblasts and endothelial cells, while a collagenase 4-based protocol failed to capture these populations despite similar overall cell recovery [52]. This highlights the critical importance of protocol customization for accurate representation of tissue ecosystems.
snRNA-Seq for Frozen and Challenging Tissues
Single-nucleus RNA sequencing (snRNA-Seq) has emerged as a powerful alternative for samples that cannot be processed as fresh tissues, including frozen archives and challenging-to-dissociate tissues [1] [52]. This approach isolates and sequences nuclei rather than whole cells, circumventing many issues associated with tissue dissociation.
The snRNA-Seq workflow involves: (1) mechanical homogenization of frozen or challenging fresh tissue to release nuclei, (2) density-based purification of nuclei, (3) nuclear viability assessment, and (4) standard droplet-based sequencing [52]. This method successfully recovers the same cell types as scRNA-Seq from matched samples, though often at different proportions due to the absence of cytoplasmic RNA [52]. snRNA-Seq is particularly valuable for tissues with extensive extracellular matrix, large cell sizes, or delicate cellular processes, such as neuronal tissues [1].
Key considerations for snRNA-Seq include the lower absolute mRNA content in nuclei compared to whole cells and the nuclear localization of specific transcripts. While snRNA-Seq captures the nuclear transcriptome effectively, it may miss biological processes regulated through cytoplasmic mRNA localization, stability, or translation [1]. Despite this limitation, studies have demonstrated successful application across diverse tissues including brain, heart, kidney, lung, pancreas, and various tumors [1] [52].
Special Considerations for FFPE Tissues
FFPE tissues represent valuable archival resources but present significant challenges for scRNA-seq due to RNA fragmentation and protein-RNA cross-linking [53]. Specialized protocols have been developed to maximize data quality from these specimens.
The FFTE workflow incorporates: (1) extended deparaffinization with xylene, (2) proteinase K digestion to reverse cross-links, (3) specialized RNA extraction protocols optimized for fragmented RNA, and (4) library preparation methods that accommodate short RNA fragments [53]. While FFPE-derived RNA is highly degraded (RIN values typically low), the DV200 metric (percentage of RNA fragments >200 nucleotides) provides a more reliable quality assessment [53].
Sequencing of FFPE-derived RNA yields higher proportions of intronic and intergenic reads compared to fresh and frozen tissues, requiring appropriate bioinformatic adjustments [53]. Despite these challenges, global gene expression profiles from FFPE tissues show high correlation with fresh and frozen tissues for protein-coding genes (ρ > 0.94), making them suitable for many research applications though with cautions regarding transcript isoform analysis [53].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for scRNA-Seq Sample Processing
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Tissue Dissociation Enzymes | Liberase TM, Collagenase IV, Papain, Pronase, Dispase, Elastase [52] | Break down extracellular matrix components; selection depends on tissue type and ECM composition [52] |
| Nuclease Inhibitors | DNase I [52] | Reduce viscosity by digesting DNA released from dead cells; improve cell viability and recovery [52] |
| RNA Stabilization Reagents | RNAlater, TRIzol, various commercial preservation solutions [53] | Preserve RNA integrity during storage and processing; critical for maintaining accurate transcriptome profiles [53] |
| Nuclei Isolation Reagents | Sucrose gradient solutions, Non-ionic detergents (e.g., NP-40), Commercial nuclei isolation kits [1] [52] | Release and purify nuclei for snRNA-Seq; maintain nuclear integrity while removing cytoplasmic contaminants [1] [52] |
| Viability Assessment Dyes | Propidium iodide, DAPI, Calcein AM, Commercial viability stains [52] | Distinguish live/dead cells or nuclei; essential for quality control before sequencing [52] |
| Library Preparation Kits | SMARTer Stranded Total RNA-Seq Kit, Commercial scRNA-seq kits (10x Genomics) [53] [1] | Convert RNA to cDNA, add barcodes, prepare sequencing libraries; optimized for low-input samples [53] [1] |
Quality Control and Data Assessment
Rigorous quality control is essential throughout the scRNA-seq workflow to ensure data reliability. For sample processing, key metrics include cell viability (recommended >80% for scRNA-Seq), successful nuclei integrity for snRNA-Seq, and RNA quality measurements [52]. The Bioanalyzer RNA Integrity Number (RIN) is valuable for fresh and frozen tissues, while the DV200 metric is more appropriate for FFPE samples [53].
Computational quality assessment should evaluate percentage of reads mapping to the transcriptome, number of genes detected per cell/nucleus, unique molecular identifier (UMI) counts, and mitochondrial read percentage [52]. Different sample types yield distinct quality metrics: FFPE samples typically show higher proportions of intronic and intergenic reads, while frozen tissues may exhibit reduced gene detection compared to fresh tissues [53]. These patterns must be considered when setting quality thresholds.
Batch effects represent a significant challenge in scRNA-seq studies, particularly when integrating data from different sample types or processing protocols. Computational batch correction tools such as Harmony, Seurat CCA, and scVI can mitigate these effects, but careful experimental design remains crucial [54]. When processing multiple samples, consistency in processing time, reagent lots, and personnel helps minimize technical variation.
The expanding toolbox for scRNA-seq sample processing has dramatically increased the range of specimens accessible for single-cell analysis. By matching processing methodologies to sample specifics—fresh tissues for maximal transcriptome coverage, frozen tissues for biobank studies, snRNA-Seq for challenging tissues, and specialized approaches for FFPE archives—researchers can extract meaningful biological insights from diverse sample types. The key principles of rapid processing for fresh tissues, appropriate stabilization for frozen specimens, and specialized protocols for difficult samples underpin successful experimental outcomes.
Emerging technologies promise to further enhance our ability to work with diverse sample types. Spatial transcriptomics approaches are bridging the gap between single-cell resolution and spatial context, allowing researchers to profile RNA transcripts within their original tissue architecture [21]. Multi-omics technologies simultaneously capture transcriptomic, epigenomic, and proteomic information from single cells, providing more comprehensive cellular characterization [11]. Computational methods continue to advance in handling the technical artifacts specific to different sample types, improving batch correction, and enabling integration of datasets generated from diverse sample sources [54].
As these technologies mature, the field moves toward increasingly standardized workflows that maintain flexibility for sample-specific adaptations. This balance between standardization and customization will be crucial for maximizing the research potential of precious clinical samples, ultimately accelerating discoveries in basic biology, disease mechanisms, and therapeutic development.
Optimizing Your scRNA-seq Experiment: Troubleshooting Common Pitfalls and Technical Challenges
A critical challenge in single-cell RNA sequencing (scRNA-seq) is the introduction of dissociation-induced artifacts—modifications of a cell's molecular profile caused by the enzymatic and mechanical processes used to create single-cell suspensions [55]. These artifacts, particularly the induction of stress-response genes, can confound data analysis and lead to inaccurate biological interpretations [56] [57]. This application note details the sources of these artifacts and provides validated wet-lab protocols and reagent solutions to mitigate them, ensuring that the transcriptional profiles obtained more faithfully represent the in vivo state.
Understanding Dissociation-Induced Stress
The process of tissue dissociation to create single-cell suspensions often requires incubation at 37°C with proteases like trypsin, collagenase, or dispase. This environment is profoundly stressful to cells, triggering rapid and artifactual changes in gene expression [56].
The most significant artifacts are the induction of immediate-early response genes (IERGs). Studies comparing dissociation methods show that traditional 37°C incubation can cause massive upregulation of genes like Fos (from 96 to over 71,000 in normalized counts) and Jun (from 3,734 to over 50,000) compared to methods designed to minimize stress [56]. Globally, over 500 genes can exhibit altered expression following a 60-minute dissociation at 37°C [56].
The table below summarizes key stress-associated genes and the quantitative impact of dissociation.
Table 1: Quantitative Impact of Dissociation on Stress Gene Expression
| Gene Category | Example Genes | Expression (Cold Protease) | Expression (37°C Protocol) | Fold-Change |
|---|---|---|---|---|
| Immediate-Early Response | Fos, Fosb, Jun, Junb | 96 - 4,834 (combined) | 15,944 - 71,082 (combined) | >15- to 70-fold [56] |
| Heat Shock / Chaperones | Hspa1a/b (HSP70) | Low / Minimal | Highly Upregulated | Significant [57] |
| Actin Cytoskeleton | Actg1, Actb | Baseline | Highly Upregulated | Significant [57] |
| Extracellular Matrix | Col1a1, Lamc1 | Baseline | Highly Upregulated | Significant [57] |
Methods for Artifact Mitigation: Protocols and Applications
Several experimental strategies have been developed to circumvent these artifacts, each with distinct advantages and applications.
Cold-Active Protease Dissociation
This method uses proteases from psychrophilic (cold-adapted) microorganisms that retain high activity at low temperatures (e.g., 6°C), where mammalian transcriptional machinery is largely inactive [56].
Experimental Protocol [56]:
- Tissue Preparation: Rapidly collect and mince tissue on a cold surface with a razor blade.
- Enzymatic Digestion: Transfer tissue to a cold-active protease solution (e.g., from Bacillus licheniformis). Incubate for 15 minutes at 6°C with gentle agitation.
- Mechanical Disruption: Augment digestion using gentle systems like the Miltenyi gentleMACS Dissociator, followed by careful trituration with a pipette.
- Filtration and Washing: Pass the cell suspension through a 30 µm filter and wash with cold buffer.
- Quality Control: Perform all steps at 6°C or colder. Assess cell viability and count before proceeding to library preparation.
Application Notes: Ideal for generating high-viability cell suspensions from tissues like kidney and developing organs. It dramatically reduces IERG induction but may be less effective for tissues with dense extracellular matrices [56] [55].
Single-Nucleus RNA Sequencing (snRNA-seq)
This approach bypasses the need for whole-cell dissociation by isolating nuclei, which are more resilient to mechanical and environmental stress [55] [58].
Experimental Protocol [55] [58]:
- Homogenization: Rapidly homogenize fresh or frozen tissue in a lysis buffer containing non-ionic detergents.
- Density Centrifugation: Purify nuclei via differential centrifugation or through a density cushion.
- Filtration and Resuspension: Filter nuclei through a suitable mesh (e.g., 20-40 µm) and resuspend in a nuclei buffer.
- Quality Control: Validate nuclei integrity and count using a fluorescent counter or microscopy.
Application Notes: snRNA-seq is the preferred method for archived frozen samples, difficult-to-dissociate tissues (e.g., brain, fibrous tissue), and when integrating with single-cell Assay for Transposase-Accessible Chromatin (scATAC-seq) for multi-omics studies [55] [58]. A key limitation is lower mRNA capture efficiency since cytoplasmic transcripts are lost [58].
Reversible Fixation (FixNCut)
This innovative method involves fixing the tissue before dissociation, effectively "freezing" the transcriptome in its native state and preventing stress-induced changes during the dissociation process [59].
Experimental Protocol [59]:
- Fixation: Incubate fresh tissue slices with a reversible cross-linker fixative, such as dithiobis(succinimidyl propionate) (DSP).
- Quenching and Washing: Quench the fixation reaction and wash the tissue thoroughly.
- Dissociation: Mechanically and enzymatically dissociate the fixed tissue.
- Reversal: Reverse the cross-links using a reducing agent (e.g., DTT) present in standard scRNA-seq reverse transcription buffers.
- Library Preparation: Proceed with standard scRNA-seq workflows.
Application Notes: FixNCut is highly robust for decentralized study designs, as sampling can be disconnected from downstream processing. It preserves RNA integrity and cellular composition while being compatible with various single-cell and spatial omics technologies [59].
Metabolic Labeling of Nascent RNA (scSLAM-seq)
This method does not prevent artifacts but provides a powerful tool to measure them, allowing for computational correction.
Experimental Protocol [57]:
- In Vivo or Ex Vivo Labeling: Incorporate 4-thiouridine (4sU) into newly synthesized RNA during the dissociation procedure.
- Cell Processing: Process the cells for scRNA-seq.
- Chemical Conversion: Treat with iodoacetamide to create T-to-C mutations in 4sU-labeled transcripts.
- Sequencing and Analysis: Identify dissociation-induced transcripts by their high T-to-C mutation rate during sequencing analysis.
Application Notes: This strategy is invaluable for optimizing dissociation protocols and for bioinformaticians to computationally separate genuine biology from technical noise [57].
The following diagram illustrates the core decision-making workflow for selecting an artifact mitigation strategy.
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for Minimizing Dissociation Artifacts
| Reagent / Solution | Function & Rationale | Example Products / Components |
|---|---|---|
| Cold-Active Protease | Enzyme with high activity at low temperatures (0-6°C), reducing transcriptional stress response. | Protease from Bacillus licheniformis [56] |
| Reversible Crosslinker | Fixes tissue transcriptome prior to dissociation; crosslinks are reversed during cDNA synthesis. | Dithiobis(succinimidyl propionate) (DSP) [59] |
| Nuclei Isolation Buffer | Lyses cytoplasmic membrane while keeping nuclei intact, enabling snRNA-seq. | Non-ionic detergents (e.g., NP-40), Sucrose, MgCl₂, Tris buffer [58] |
| RNA Labeling Nucleoside | Incorporated into newly transcribed RNA, allowing identification of dissociation-induced transcripts. | 4-thiouridine (4sU) [57] |
| Chemical Conversion Reagent | Alkylates 4sU-labeled RNA, causing T-to-C mutations in sequencing reads for detection. | Iodoacetamide (IAA) [57] |
| Transcriptional Inhibitors | Chemicals that block RNA polymerase, potentially halting stress response (use with caution due to side effects). | Actinomycin D [55] |
Comparative Analysis of Method Performance
The choice of method involves trade-offs between data quality, practicality, and compatibility with downstream assays. The following diagram and table provide a high-level comparison of the primary methods discussed.
Table 3: Comparison of Artifact Mitigation Methods
| Method | Key Advantage | Key Limitation | Best Suited For |
|---|---|---|---|
| Cold-Active Protease | Preserves high-quality transcriptome from intact cells; minimizes IERG induction [56]. | May be ineffective for tough tissues; requires optimization of cold-digestion [55]. | Standard scRNA-seq on fresh, readily dissociable tissues. |
| Single-Nucleus RNA-seq | Bypasses dissociation stress entirely; applicable to frozen and difficult tissues [55] [58]. | Loss of cytoplasmic mRNA reduces genes detected per cell [58]. | Biobank samples, complex tissues (brain, tumor stroma), multiome studies. |
| Reversible Fixation (FixNCut) | Decouples sampling from processing; locks in transcriptome prior to stress [59]. | Protocol complexity; potential for incomplete reversal of crosslinks [59]. | Multi-site studies, clinical biopsies, and spatial transcriptomics integration. |
| RNA Labeling (scSLAM-seq) | Empowers computational correction by quantifying artifact transcripts [57]. | Does not prevent artifacts; adds experimental and computational steps [57]. | Protocol optimization and studies where computational correction is preferred. |
The success of single-cell RNA sequencing (scRNA-seq) hinges on the initial quality of the single-cell suspension. Inadequate tissue dissociation can introduce technical artifacts, skew cellular composition, and compromise the integrity of the entire dataset [60]. Achieving a balance between high cell yield, viability, and representative sampling of all original cell populations is a significant challenge. This application note details the current best practices in enzymatic and mechanical dissociation, providing a structured guide for researchers to optimize this critical first step in the scRNA-seq workflow.
The State of Tissue Dissociation: Methods and Trade-offs
Tissue dissociation requires breaking down the extracellular matrix (ECM) and cell-cell junctions. Traditional methods combine mechanical and enzymatic approaches, but newer technologies offer innovative alternatives.
The table below summarizes the performance of various dissociation technologies as reported in recent literature:
Table 1: Comparison of Modern Tissue Dissociation Technologies
| Technology | Dissociation Type | Tissue Type (Example) | Cell Viability | Time | Key Efficacy Findings |
|---|---|---|---|---|---|
| Optimized Chemical-Mechanical [60] | Enzymatic & Mechanical | Bovine Liver | >90% | 15 min | 92% ± 8% dissociation efficacy (vs. 37-42% enzymatic only) |
| Cold-Active Protease [61] | Enzymatic (Cold) | Mouse Kidney | Varies by cell type | ~1 hour | Minimizes stress gene expression; better preserves sensitive cells (e.g., podocytes) |
| Warm Enzymatic (37°C) [61] | Enzymatic (Warm) | Mouse Kidney | Varies by cell type | ~1 hour | Induces stress response genes; can underrepresent fragile cell types |
| Hypersonic Levitation (HLS) [62] | Ultrasound (Non-contact) | Human Renal Cancer | 92.3% | 15 min | 90% tissue utilization; excels at preserving rare cell populations |
| Microfluidic Platform [60] | Microfluidic & Enzymatic | Mouse Kidney, Breast Tumor | 60%-95% (varies by cell type) | 20-60 min | ~20,000 cells/mg tissue (kidney epithelial cells) |
Experimental Protocols for Robust Single-Cell Dissociation
Optimized Enzymatic-Mechanical Dissociation for scRNA-seq
The following protocol, adapted from recent studies on skin and other tissues, is designed to minimize transcriptional stress and maximize viability [63] [61].
Reagents and Equipment:
- Dulbecco’s Phosphate Buffered Saline (DPBS)
- Collagenase IV: Degrades collagen in the ECM.
- Dispase II: A neutral protease that cleaves fibronectin and collagen IV, gentler on cell membranes.
- DNase I: Prevents cell clumping by digesting free DNA released from damaged cells.
- Fetal Bovine Serum (FBS): Used to inactivate enzymes and stop digestion.
- Cell Strainers (40 µm and 70 µm)
- Orbital Shaker or Shaking Water Bath
Table 2: Reagent Toolkit for Enzymatic Dissociation
| Reagent/Equipment | Function | Considerations for scRNA-seq |
|---|---|---|
| Collagenase IV [63] | Digests interstitial collagen | Preferred over harsher proteases like trypsin to preserve surface markers. |
| Dispase II [63] | Cleaves basement membrane proteins | Often used in combination with collagenase for more complete dissociation. |
| DNase I [63] | Degrades DNA | Critical for reducing viscosity from lysed cells, preventing clumping. |
| Cold-Active Protease [61] | Functions at low temperatures (e.g., on ice) | Minimizes artifactual stress-induced gene expression during dissociation. |
| EDTA [60] | Chelating agent | Disrupts cell-cell adhesions by binding calcium. |
Step-by-Step Protocol:
- Tissue Collection and Transport: Place the fresh tissue sample (e.g., a 4 mm punch biopsy) in complete medium (e.g., RPMI with 10% FBS) and store at 4°C. Process within 2 hours of collection [63].
- Initial Mechanical Disruption:
- Transfer tissue to a culture dish with a small volume of dissociation buffer.
- Using a sterile scalpel, mince the tissue into fine fragments (approximately 1-2 mm³).
- Enzymatic Digestion:
- Transfer the minced tissue into a tube containing pre-warmed enzyme solution. A typical combination is 2 mg/mL Collagenase IV and 1 mg/mL Dispase in DPBS, with 0.1 mg/mL DNase I [63].
- For sensitive transcriptomic applications, perform digestion on ice using cold-active protease to avoid inducing a stress response [61].
- Incubate the tube on an orbital shaker (or in a shaking water bath) at 80-100 rpm. Digestion time typically ranges from 30 minutes to 2 hours, depending on tissue density.
- Termination and Filtration:
- Quench the enzymatic reaction by adding a volume of cold FBS or complete medium equivalent to at least half the digestion volume.
- Pipette the cell suspension up and down gently to mechanically assist dissociation.
- Pass the suspension through a 70 µm cell strainer, followed by a 40 µm cell strainer, to remove undigested tissue and large aggregates.
- Cell Washing and Counting:
- Centrifuge the filtrate and resuspend the cell pellet in a suitable buffer (e.g., DPBS with 0.04% BSA).
- Count cells and assess viability using an automated cell counter with AO/PI staining or Trypan Blue exclusion.
Workflow for Method Selection and Execution
The following diagram illustrates the key decision points in the tissue dissociation workflow, guiding researchers toward an optimal protocol.
Diagram 1: A decision workflow for selecting an appropriate tissue dissociation method based on research priorities and tissue type.
Key Considerations for Protocol Optimization
- Temperature is Critical: Digestion at 37°C (warm dissociation) significantly induces immediate-early genes (e.g., FOS, JUN) and heat shock proteins, creating transcriptomic artifacts [61]. Cold dissociation on ice is highly recommended for scRNA-seq to capture a more biologically accurate state [61].
- Cell Type Biases: Different dissociation methods can enrich or deplete specific cell types. Warm dissociation, for instance, can lead to a severe underrepresentation of fragile cells like podocytes and endothelial cells [61]. The choice of method should be validated for the cell populations of interest.
- Emerging Technologies: Non-contact methods like Hypersonic Levitation and Spinning (HLS) show great promise by using acoustic energy to levitate and dissociate tissue with high efficiency and viability while better preserving rare cell types [62]. For formalin-fixed paraffin-embedded (FFPE) tissues, cryogenic enzymatic dissociation (snCED-seq) offers a robust solution for nuclei isolation [64].
A one-size-fits-all approach does not exist for tissue dissociation in single-cell research. The optimal protocol is a careful balance between dissociation efficiency, cell viability, and transcriptional fidelity. By understanding the trade-offs between enzymatic, mechanical, and emerging non-contact methods, researchers can make informed decisions to ensure their single-cell suspensions provide a solid foundation for groundbreaking discoveries.
Obtaining high-quality single-cell suspensions is a critical, yet often limiting, step in single-cell RNA sequencing (scRNA-seq) workflows. The process is particularly challenging for sensitive tissues and those with high intrinsic RNase activity, such as adipose tissue, pancreas, and spleen [65] [66]. Maintaining RNA integrity during sample preparation is paramount for accurately capturing the transcriptome, as degradation can introduce significant technical noise and bias, compromising data quality [65]. This application note details optimized protocols and key considerations for mitigating these challenges, ensuring high cell viability and yield for reliable scRNA-seq results.
Key Challenges and Tissue-Specific Considerations
The approach to tissue dissociation must be tailored to the specific tissue of interest, as cellular architecture, extracellular matrix (ECM) composition, and intrinsic RNase levels vary significantly.
Table 1: Tissue-Specific Dissociation Challenges and Strategies
| Tissue Type | Key Challenges | Recommended Strategies |
|---|---|---|
| Adipose Tissue | Large, fragile lipid-filled adipocytes; high RNase activity [66]. | Use single-nucleus RNA-seq (snRNA-seq) to bypass cell size issues [66]. Incorporate robust RNase inhibitors like VRC [66]. |
| Brain | Intricate neuronal structures; myelin sheath [67]. | Use snRNA-seq for large neurons; include a myelin removal step in droplet-based protocols [67]. |
| Tumors | Fibrous/calcified regions; necrotic areas; high cellular density and cohesion [67]. | Use validated, tissue-specific enzymatic cocktails; combine mechanical and enzymatic methods [67]. |
| iPS Cell Colonies | Densely packed colonies; strong cell-cell adhesion [67]. | Use enzymes targeting specific adhesion molecules; optimize incubation time to prevent over-dissociation [67]. |
The decision between analyzing single cells or single nuclei is fundamental. scRNA-seq captures a comprehensive profile of both nuclear and cytoplasmic RNA, making it ideal for studying splicing and cytoplasmic genes. However, snRNA-seq is often necessary for tissues that are difficult to dissociate (e.g., fibrous tissues), for very large cells (e.g., adipocytes, neurons), or when working with frozen or archived tissue samples [67] [66].
Optimized Dissociation and Handling Protocols
General Workflow for Single-Cell Suspension Preparation
The following diagram outlines the core decision points and steps for preparing a high-quality single-cell suspension.
Detailed Protocol for Enzymatic Dissociation of Challenging Tissues
This protocol is adapted for RNase-rich and fibrous tissues [67] [68].
Before You Begin:
- Pre-cool all buffers and centrifuges to 4°C.
- Thaw enzymes on ice and pre-warm dissociation medium to 37°C.
- Turn on the cell culture hood 15 minutes prior.
Step-by-Step Method:
- Tissue Collection and Mincing:
- Immediately after harvest, place the tissue in a Petri dish with cold, sterile PBS or HBSS.
- Using sterile scalpels or razor blades, mince the tissue into fine pieces (approximately 1-2 mm³).
Enzymatic Digestion:
- Transfer the minced tissue to a tube containing pre-warmed dissociation medium. A recommended base recipe is Hank's Balanced Salt Solution (HBSS) supplemented with 0.5 mg/mL Collagenase Type II and 0.04% Bovine Serum Albumin (BSA) [68].
- Incubate the tube at 37°C for 20-45 minutes on an orbital shaker with gentle agitation. The incubation time must be optimized for each tissue type.
Mechanical Dissociation:
- Periodically during enzymatic incubation, triturate the tissue mixture 10-15 times using a sterile serological pipette or a fire-polished Pasteur pipette.
- For tougher tissues, a gentle mechanical homogenizer (e.g., gentleMACS Dissociator) can be used.
Reaction Termination and Filtration:
- Add an equal volume of cold HBSS with 4% BSA to neutralize the enzymes.
- Pass the cell suspension through a pre-wet 40 µm cell strainer into a new tube to remove undigested tissue and large clumps [68].
Washing and Resuspension:
Protocol for Single-Nucleus Isolation from RNase-Rich Tissues
This protocol is optimized for tissues like adipose and liver, where RNA degradation is a major concern [66].
Homogenization:
- Place minced tissue (up to 0.5 cm³) in a Dounce homogenizer containing 2-5 mL of ice-cold lysis buffer. The optimized lysis buffer should contain:
- Vanadyl Ribonucleoside Complex (VRC) at a recommended concentration (e.g., 10-20 mM) to preserve RNA integrity [66].
- Recombinant RNase Inhibitor (RRI) (e.g., 0.4 U/µL).
- Dounce the tissue 15-25 times with the loose pestle (A), then 10-15 times with the tight pestle (B), all performed on ice.
- Place minced tissue (up to 0.5 cm³) in a Dounce homogenizer containing 2-5 mL of ice-cold lysis buffer. The optimized lysis buffer should contain:
Nuclei Purification and Washing:
- Filter the homogenate through a 40 µm strainer into a new tube.
- Underlay the filtrate with a cushion of sucrose solution or density gradient medium.
- Centrifuge at 500-1000 x g for 10-20 minutes at 4°C to pellet the nuclei.
- Resuspend the purified nuclei pellet in a wash buffer with RRI and VRC.
Quality Control and Storage:
- Count nuclei using an automated counter or hemocytometer.
- Nuclei isolated with this optimized protocol can maintain RNA integrity for up to 24 hours at 4°C, enabling greater experimental flexibility [66].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for scRNA-seq Sample Prep
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Collagenase Type II | Enzymatic dissociation of tissues rich in collagen (e.g., tumors, connective tissues) [67]. | Different types (I, II, IV) are optimized for different tissues; concentration and time require optimization [67]. |
| TrypLE | Gentle enzymatic alternative to trypsin for dissociating adherent cell lines and cultures [67]. | Less toxic than trypsin; ideal for sensitive primary cells [67]. |
| Vanadyl Ribonucleoside Complex (VRC) | Potent RNase inhibitor that protects nuclear RNA during isolation from challenging tissues [66]. | Superior to RRIs alone for tissues like adipose and liver; note potential inhibition of reverse transcriptases [66]. |
| Synthetic Thermostable RNase Inhibitor (e.g., SEQURNA) | Thermostable, non-protein-based inhibitor for use during cell lysis and reverse transcription [65]. | Retains activity during heat denaturation steps (e.g., 72°C), improving workflow flexibility and reproducibility [65]. |
| Hank's Balanced Salt Solution (HBSS) | Salt solution used as a base for enzymatic dissociation, providing ions for enzyme function [68]. | Pre-warm to 37°C for optimal enzymatic activity during digestion. |
| Mg2+/Ca2+-Free PBS | Buffer for washing and resuspending cells post-digestion and for FACS sorting [69]. | Prevents clumping and avoids interference with downstream enzymatic steps like reverse transcription [69]. |
| 40 µm Cell Strainer | Removes undigested tissue fragments and cell clumps to ensure a single-cell suspension [68]. | Critical for preventing microfluidic channel clogging in droplet-based platforms. |
Critical Factors for Success
- Temperature Control: Perform dissociation steps at cold temperatures where possible to slow RNA degradation and metabolic activity. However, note that enzymatic activity is optimal at 37°C, requiring a balanced approach [67].
- Cell Viability: The presence of excessive dead cells can negatively impact scRNA-seq data. Use viability dyes like propidium iodide (PI) or trypan blue for accurate assessment. Combining mechanical and enzymatic methods can help minimize cell death [67].
- RNase Inhibition: For RNase-rich tissues, standard recombinant RNase inhibitors may be insufficient. The combination of VRC and RRI has been demonstrated to significantly improve RNA integrity in the most challenging contexts, such as visceral adipose tissue [66].
- Speed and Cold Chain: Work quickly and keep samples on ice throughout the process to minimize RNA degradation and stress-induced gene expression changes [69].
Addressing Amplification Bias and Low mRNA Capture Efficiency
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the characterization of gene expression profiles at the level of individual cells, revealing cellular heterogeneity that is masked in bulk sequencing approaches [21] [70]. However, two significant technical challenges persistently compromise data quality and biological interpretation: amplification bias and low mRNA capture efficiency. These issues are particularly problematic when working with clinical samples or rare cell populations where material is limited [71]. Amplification bias during polymerase chain reaction (PCR) can dramatically skew transcript abundance measurements, while low mRNA capture efficiency results in sparse data with excessive zeros, complicating downstream analysis [72] [73]. This Application Note provides detailed protocols and analytical frameworks to address these challenges, ensuring more accurate and reproducible single-cell transcriptomic data within the broader context of developing robust single-cell RNA sequencing protocols for isolates research.
Understanding Amplification Bias in scRNA-seq
Mechanisms and Impact of Amplification Bias
In multi-template PCR, a critical step in most scRNA-seq protocols, different DNA templates amplify with varying efficiencies due to sequence-specific characteristics. This non-homogeneous amplification results in skewed abundance data that no longer reflects original transcript proportions [73]. Even minor differences in amplification efficiency (as little as 5% below average) can cause substantial under-representation of certain transcripts after just 12 PCR cycles, with severely affected sequences becoming virtually undetectable after 60 cycles [73]. This bias compromises the accuracy and sensitivity of quantitative results, potentially leading to erroneous biological conclusions.
Recent research has identified that sequence-specific factors beyond traditional culprits like GC content contribute significantly to amplification bias. Specific motifs adjacent to adapter priming sites, particularly those facilitating adapter-mediated self-priming, have been identified as major mechanisms causing low amplification efficiency [73].
Quantitative Assessment of Amplification Bias
Table 1: Impact of PCR Cycles on Sequence Coverage Dropout
| Number of PCR Cycles | Fraction of Sequences with Severe Depletion | Fold Reduction in Recovery Rate |
|---|---|---|
| 15 | <1% | 1.5x |
| 30 | ~2% | 3x |
| 45 | ~5% | 6x |
| 60 | >8% | >10x |
Data derived from serial amplification experiments on synthetic oligonucleotide pools [73]
Experimental Protocols for Bias Mitigation
Protocol: Deep Learning-Assisted Amplicon Library Design
This protocol utilizes computational prediction to design amplicon libraries with inherently homogeneous amplification properties [73].
Materials:
- Synthetic DNA pool with defined sequences
- One-dimensional convolutional neural network (1D-CNN) model
- Standard PCR reagents
- Next-generation sequencing platform
Procedure:
- Sequence Analysis: Input candidate amplicon sequences into a trained 1D-CNN model to predict sequence-specific amplification efficiencies
- Library Optimization: Select sequences with predicted high and uniform amplification efficiencies for experimental use
- Validation: Perform small-scale serial amplification (15, 30, 45 cycles) on synthetic pool to verify homogeneous coverage
- Sequencing: Library preparation and sequencing across multiple PCR cycles
- Quality Control: Calculate coverage distributions and confirm reduced skewness compared to non-optimized libraries
Expected Outcomes: This approach reduces the required sequencing depth to recover 99% of amplicon sequences fourfold and significantly decreases sequence dropout rates [73].
Protocol: Unique Molecular Identifier (UMI) Integration
UMIs are short random sequences used to label individual mRNA molecules during reverse transcription, enabling correction of amplification bias during computational analysis [70].
Materials:
- Poly(T) primers with integrated UMI sequences
- Reverse transcription reagents
- PCR amplification reagents
- Standard bioinformatics pipeline for UMI counting
Procedure:
- Cell Lysis: Perform in single-cell lysis buffer
- Reverse Transcription: Use custom primers containing cell barcode, UMI, and adapter sequences
- cDNA Amplification: Perform limited-cycle PCR
- Library Preparation: Follow standard protocols for your platform
- Computational Analysis: Count UMIs rather than reads to quantify transcript abundance
Troubleshooting: Ensure UMI diversity is sufficient to minimize collision probability. Use at least 4^N possible UMIs where N is UMI length.
Addressing Low mRNA Capture Efficiency
Understanding mRNA Capture Challenges
Low mRNA capture efficiency in scRNA-seq manifests as a high proportion of zero counts in the expression matrix, potentially representing: (1) genuine biological zeros indicating no expression; (2) sampled zeros from lowly expressed genes; or (3) technical zeros from failed capture despite expression [72]. The prevailing notion that zeros are largely technical artifacts has led to preprocessing steps that may discard biologically meaningful information, particularly for rare cell types or low-abundance transcripts [72].
Protocol choice significantly impacts capture efficiency. Full-length methods like Smart-Seq2 detect more genes per cell, while 3' end-counting methods like droplet-based platforms offer higher throughput [11] [70]. Single-nuclei RNA sequencing (snRNA-seq) presents an alternative for samples where tissue dissociation is challenging, though it typically yields lower RNA content compared to whole-cell approaches [58].
Quantitative Comparison of Platform Efficiencies
Table 2: Capture Efficiencies Across Commercial scRNA-seq Platforms
| Platform | Technology | Capture Efficiency | Cell Size Limit | Recommended Application |
|---|---|---|---|---|
| 10× Genomics Chromium | Microfluidic droplets | 70-95% | 30 µm | High-throughput cell typing |
| BD Rhapsody | Microwell partitioning | 50-80% | 30 µm | Targeted transcript detection |
| Parse Evercode | Multiwell-plate | >90% | No strict limit | High-cell number studies |
| Fluidigm C1 | Microfluidic chambers | 60-85% | 25 µm | Full-length transcript analysis |
| Singleron SCOPE-seq | Microwell partitioning | 70-90% | <100 µm | Large or specialized cells |
Data compiled from multiple sources [58] [71] [70]
Integrated Workflow for Comprehensive Bias Mitigation
Integrated scRNA-seq Bias Mitigation Workflow
Advanced Statistical Approaches for Data Correction
GLIMES Framework for Zero-Inflation and Batch Effects
The GLIMES (Generalized Linear Mixed Effects for Single-cell) statistical framework addresses multiple challenges in scRNA-seq analysis by leveraging UMI counts and zero proportions within a generalized Poisson/Binomial mixed-effects model [72].
Implementation Protocol:
- Input Data Preparation: Use raw UMI counts without imputation or excessive zero-filtering
- Model Specification: Include random effects for donor/batch variations and fixed effects for experimental conditions
- Parameter Estimation: Implement maximum likelihood estimation with regularization
- Differential Expression Testing: Compute p-values with multiple testing correction
Advantages: GLIMES preserves biologically meaningful signals by using absolute RNA expression rather than relative abundance, improving sensitivity and reducing false discoveries compared to methods that rely on normalized data [72].
Benchmarking Performance
In rigorous benchmarking against six existing differential expression methods, GLIMES demonstrated superior adaptability to diverse experimental designs and more effective mitigation of key shortcomings, particularly those related to normalization procedures [72].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Bias Mitigation
| Reagent/Method | Function | Example Products/Protocols |
|---|---|---|
| Unique Molecular Identifiers (UMIs) | Corrects for amplification biases | 10× Barcoded beads, Parse Biosciences |
| Template-Switching Oligos | Enhances cDNA synthesis completeness | Smart-Seq2, SMARTer kits |
| Degenerate Primers | Reduces sequence-specific bias | Custom-designed pools |
| Multiplex PCR Kits | Enables targeted amplification | Mission Bio Tapestri |
| Spike-in RNA Controls | Monitors technical variation | ERCC RNA Spike-In Mix |
| Fixation Reagents | Preserves cell state for complex samples | Glyoxal, Paraformaldehyde |
| Nuclei Isolation Kits | Enables sequencing from difficult tissues | 10× Nuclei Isolation Kits |
Information compiled from multiple sources [58] [71] [41]
Amplification bias and low mRNA capture efficiency remain significant challenges in single-cell RNA sequencing, but integrated experimental and computational approaches can substantially mitigate their impact. By implementing sequence-aware library design, utilizing UMIs, selecting appropriate platforms, and applying advanced statistical frameworks like GLIMES, researchers can achieve more accurate and biologically meaningful results. These protocols provide a foundation for robust single-cell RNA sequencing of isolates, particularly valuable for clinical samples and rare cell populations where material is precious and technical artifacts can most easily obscure biological truth.
Mitigating Batch Effects and Controlling for Confounding Technical Variation
In single-cell RNA sequencing (scRNA-seq) research, batch effects represent technical variations arising from processing samples at different times, with different protocols, reagents, or personnel [11]. These non-biological variations can confound biological signals, leading to inaccurate cell type identification, erroneous differential expression results, and compromised data integration [74] [75]. As scRNA-seq moves toward large-scale atlas projects integrating datasets from multiple laboratories and platforms, mitigating these effects has become a critical prerequisite for robust biological insight [76]. This application note provides a comprehensive framework for identifying, controlling, and correcting for technical variation throughout the scRNA-seq workflow, from experimental design to computational analysis.
Experimental Design and Wet-Lab Protocols
Strategic Experimental Design for Batch Effect Minimization
Proactive experimental design is the most effective strategy for batch effect control. Randomization of sample processing order across experimental groups prevents confounding between biological conditions and technical batches. Whenever feasible, incorporating technical replicates – where the same biological sample is processed in multiple batches – provides direct estimation of technical variation. For project designs involving multiple samples processed over time, implementing blocking strategies, where each batch contains cells from all experimental conditions, enables statistical separation of biological and technical effects [11].
For studies comparing primary and metastatic tumors, as in a recent ER+ breast cancer investigation, standardizing processing protocols across all samples is paramount. This includes using identical tissue dissociation methods, single-cell suspension generation, and library construction protocols to minimize introduced variability [77].
Optimized Single Nuclei Isolation Protocol for Clinical Biopsies
For samples where fresh processing is impractical or tissues are difficult to dissociate, single nuclei RNA-seq (snRNA-seq) provides a robust alternative. The following optimized protocol for frozen clinical biopsies eliminates sorting and ultra-centrifugation, processing samples in approximately 90 minutes while preserving nuclei integrity [78].
Table: Reagents and Equipment for Single Nuclei Isolation
| Item | Specification | Function |
|---|---|---|
| Lysis Buffer | 1× PBS pH 7.4, 10 mM Tris-HCL, 0.0125% Triton X-100, 1 mM DTT, 0.2 U/μL RNAse inhibitor | Disrupts cellular membranes while preserving nuclear integrity |
| Wash/Resuspension Buffer | 1× PBS pH 7.4, 2% BSA, 0.2 U/μL RNAse inhibitor | Stabilizes nuclei and inhibits RNA degradation |
| Centrifuge | Swinging bucket rotor, pre-cooled to 4°C | Pellet nuclei while minimizing mechanical damage |
| Cell Strainers | 70 μM and 40 μM FlowMi filters | Remove aggregates and debris |
| DAPI Staining Solution | - | Nuclei quantification and viability assessment |
Protocol Steps [78]:
- Preparation: Freshly prepare all reagents, decontaminate equipment with RNAsin, and pre-chill centrifuges to 4°C. Pre-coat tips and tubes with 5% BSA to reduce nuclei retention.
- Tissue Processing: Remove needle biopsies from -80°C storage and mince to 0.5-1 mm pieces on dry ice using a sterile scalpel blade.
- Mechanical Lysis: Transfer tissue to 2 mL tube containing cold lysis buffer and a micro stir-rod. Place on magnetic stir plate at 100 RPM for 5 minutes on ice.
- Supernatant Collection and Washing: After lysis, transfer supernatant to 15 mL tube containing 6 mL wash buffer. Repeat lysis and washing steps twice more for higher nuclei yield.
- Centrifugation and Filtration: Centrifuge at 600×g for 5 minutes at 4°C. Resuspend pellet in wash buffer, then sequentially filter through 70 μM and 40 μM strainers.
- Quantification: Count nuclei using DAPI staining and hemocytometer or automated cell counter. Assess viability with Trypan blue exclusion.
This protocol has demonstrated effectiveness in obtaining normal nuclei morphology without structural damage across multiple tissue types, including human kidney, heart, liver, and brain tissues [78].
Single Nuclei Isolation Workflow from Frozen Clinical Biopsies
Computational Correction Methods
Comprehensive Comparison of Batch Correction Tools
When batch effects persist despite optimal experimental design, computational correction methods are essential. Recent benchmarking studies evaluating eight widely used methods revealed significant differences in performance and potential artifacts introduced during correction [74] [75].
Table: Comparative Analysis of scRNA-seq Batch Correction Methods
| Method | Underlying Algorithm | Input Data | Correction Approach | Performance Assessment | Key Considerations |
|---|---|---|---|---|---|
| Harmony | Soft k-means with linear correction | Normalized counts | Corrects embedding using soft clustering | Consistently performs well without introducing artifacts [75] | Preserves biological variation while removing technical effects |
| ComBat/ComBat-Seq | Empirical Bayes framework | Raw/Normalized counts | Linear correction of count values | Introduces detectable artifacts in some setups [75] | pyComBat offers Python implementation with faster computation [79] |
| SCVI | Conditional variational autoencoder | Raw count matrix | Learns low-dimensional latent space | Performs poorly, often altering data considerably [75] | Struggles with substantial batch effects across systems [76] |
| Seurat | Canonical Correlation Analysis (CCA) | Normalized counts | Aligns canonical basis vectors | Introduces artifacts in correction process [75] | Popular in many workflows but requires careful validation |
| BBKNN | Graph-based correction | k-NN graph | Modifies neighborhood graph | Introduces detectable artifacts [75] | Fast for large datasets but changes graph structure |
| MNN | Mutual nearest neighbors | Normalized counts | Linear correction based on MNN pairs | Performs poorly, alters data considerably [75] | Early method superseded by more robust approaches |
| LIGER | Quantile alignment | Normalized counts | Aligns factor loadings | Performs poorly, often altering data considerably [75] | Tends to over-correct and remove biological variation |
| sysVI | VampPrior + cycle-consistency | Normalized counts | Conditional VAE with consistency constraints | Effective for substantial batch effects [76] | Newer method for challenging integration scenarios |
Implementation Guidelines for Computational Correction
Based on recent benchmarking studies, Harmony currently represents the most reliably calibrated method, effectively removing batch effects while minimizing introduction of artifacts [75]. The algorithm employs soft k-means clustering with linear correction within cluster domains, operating on normalized count matrices and returning a corrected embedding without altering the original count structure.
For Python-based workflows, pyComBat provides a efficient implementation of the established ComBat algorithm, demonstrating 4-5 times faster computation than the original R implementation while producing equivalent correction results [79]. The tool supports both parametric (faster) and non-parametric (more robust to distributional assumptions) approaches.
For exceptionally challenging integration scenarios involving substantial batch effects across different systems (e.g., cross-species, organoid-to-tissue, or single-cell to single-nuclei), newer methods like sysVI show promise. This approach combines variational inference with cycle-consistency constraints to handle strong technical and biological confounders while preserving relevant biological variation [76].
Batch Effect Correction Strategy Selection Framework
Quality Control and Validation Metrics
Pre- and Post-Correction Assessment Framework
Rigorous quality control is essential both before and after batch correction implementation. Pre-correction assessment should evaluate batch effect strength using metrics like Principal Variance Component Analysis (PVCA) to quantify the proportion of variance attributable to batch versus biological factors. The iLISI (graph integration local inverse Simpson's index) metric specifically measures batch mixing in local neighborhoods, with scores approaching 1 indicating optimal batch integration [76].
Post-correction validation must confirm that biological variation has been preserved while technical artifacts have been removed. This includes visualizing cell-type clustering consistency across batches using UMAP or t-SNE embeddings, and verifying that known biological groups (e.g., cell types from different conditions) remain separable while technical replicates merge appropriately. Differential expression analysis between batches should yield minimal significant genes after successful correction [75].
Case Study: Batch Effect Management in Breast Cancer Research
In a recent scRNA-seq study of ER+ breast cancer comparing primary and metastatic tumors, researchers implemented a comprehensive batch effect control strategy. They processed all samples using standardized dissociation and library preparation protocols, incorporated sample identity as a covariate in their integration model (SCVI), and validated correction by confirming that expected biological differences in malignant cells and tumor microenvironment components were preserved while technical variability was minimized [77]. The study successfully identified distinct transcriptional states between primary and metastatic cells and characterized specific immune cell subtypes enriched in metastatic microenvironments, demonstrating effective separation of biological signals from technical noise.
Advanced Applications and Specialized Tools
Causal Inference for Perturbation Response Disentanglement
For perturbation studies (e.g., drug treatments, genetic manipulations), distinguishing true treatment effects from pre-existing cellular heterogeneity requires specialized causal inference approaches. Tools like scCausalVI implement structural causal models to disentangle intrinsic cellular states from extrinsic perturbation effects through cross-condition in silico prediction [80]. This methodology enables researchers to computationally predict how individual cells would respond under alternative conditions, addressing the fundamental limitation of single-cell destruction during measurement.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table: Key Research Reagent Solutions for Batch Effect Control
| Reagent/Tool | Function | Application Context | Considerations |
|---|---|---|---|
| RNAlater Preservation Solution | Stabilizes RNA in fresh tissues | Biobanking clinical biopsies collected at unpredictable times | Enables snRNA-seq from archived samples [78] |
| Triton X-100 Detergent | Cellular membrane disruption | Nuclei isolation lysis buffer (0.0125% concentration) | Optimized concentration preserves nuclear integrity [78] |
| RNAse Inhibitors | Prevents RNA degradation | All steps from tissue processing to library preparation | Critical for maintaining RNA quality in low-input protocols |
| BSA (Bovine Serum Albumin) | Blocks non-specific binding | Coating tips/tubes; wash buffer component (2% concentration) | Significantly improves nuclei recovery [78] |
| DAPI Stain | DNA intercalating dye | Nuclei quantification and viability assessment | Distinguishes intact nuclei from debris [78] |
| Unique Molecular Identifiers (UMIs) | Corrects PCR amplification bias | Most modern scRNA-seq protocols | Essential for accurate transcript quantification [11] |
| Cell Hashing Oligos | Sample multiplexing | Pooling multiple samples in single run | Redances batch effects and costs [11] |
Effective management of batch effects and technical variation requires an integrated approach spanning experimental design, wet-lab protocols, and computational correction. Standardization of sample processing, implementation of appropriate control strategies, and selection of carefully calibrated computational methods form the foundation of robust scRNA-seq research. As the field progresses toward increasingly complex multi-sample atlas projects, the rigorous application of these principles will be essential for extracting biologically meaningful insights from single-cell transcriptomic data.
Validating and Interpreting scRNA-seq Data: From Computational Analysis to Biological Confirmation
Single-cell RNA sequencing (scRNA-seq) has revolutionized transcriptomic research by enabling the exploration of gene expression at the level of individual cells, uncovering cellular heterogeneity that is often masked in bulk RNA-seq analyses [21]. Since its inception in 2009, scRNA-seq has evolved into a pivotal tool for studying complex biological systems, from developmental biology to tumor microenvironments [81] [71]. The technology has motivated the development of hundreds of computational methods designed to interpret the unique data structures it generates [81]. However, the analysis of scRNA-seq data presents distinct challenges due to its high-dimensional, sparse, and noisy nature, necessitating specialized bioinformatics pipelines [70]. This document outlines essential bioinformatics workflows for scRNA-seq data analysis, focusing specifically on quality control, normalization, and clustering—three fundamental steps that form the foundation for all downstream biological interpretations. Framed within the context of a broader thesis on single-cell RNA sequencing protocols for isolated cells, this guide provides detailed methodologies and best practices to ensure robust, reproducible analysis for researchers, scientists, and drug development professionals.
Single-Cell RNA Sequencing Workflow: From Cells to Clusters
The journey from a biological sample to meaningful cluster assignments involves a multi-step process, each stage critical for ensuring data integrity. The following diagram illustrates the primary workflow in a typical scRNA-seq bioinformatics pipeline.
Phase 1: Quality Control (QC)
Objectives and Rationale
Quality control is the first and most critical phase in scRNA-seq data analysis, aimed at distinguishing high-quality cells from low-quality ones and technical artifacts. The primary goal is to eliminate barcodes representing empty droplets, dead cells, damaged cells, or multiple cells (multiplets) that could confound downstream biological interpretations [82]. Effective QC minimizes technical noise while preserving biological signal, ensuring that subsequent analyses reflect true biology rather than experimental artifacts. As scRNA-seq data are susceptible to various technical confounders, including batch effects, amplification biases, and ambient RNA contamination, rigorous QC practices are non-negotiable for generating reliable results [70].
Key QC Metrics and Interpretation
The table below summarizes the core metrics used to assess cell quality in scRNA-seq experiments, their biological or technical interpretations, and recommended filtering thresholds.
Table 1: Essential Quality Control Metrics for scRNA-seq Data
| QC Metric | Description | Biological/Technical Meaning | Recommended Thresholds |
|---|---|---|---|
| UMI Counts | Total number of Unique Molecular Identifiers per cell | Measures sequencing depth; low counts may indicate empty droplets, high counts may suggest multiplets | Filter extreme outliers in distribution [82] |
| Genes Detected | Number of genes detected per cell | Indifies library complexity; correlates with UMI counts | Filter extreme outliers in distribution [82] |
| Mitochondrial Read Percentage | Percentage of reads mapping to mitochondrial genes | Indicator of cell stress or damage; high percentages suggest low viability | <10% for PBMCs; cell-type dependent [82] |
| Cell Recovery | Number of cells recovered versus targeted | Assesses technical efficiency of experiment | Varies by platform (e.g., ~80% for Chromium X) [12] |
Practical Protocol: Implementing Quality Control
Initial Assessment with Cell Ranger Output
For data generated from 10x Genomics platforms, begin by examining the web_summary.html file generated by the Cell Ranger pipeline. This report provides a comprehensive overview of key quality metrics, including:
- Confirmed cell count and estimated number of cells
- Mean reads per cell and sequencing saturation
- Fraction of reads confidently mapped to the transcriptome
- Median genes detected per cell
- Median UMI counts per cell [82]
The summary file will flag critical issues with a warning message. A successful run typically displays a message stating "No critical issues were identified in this analysis" and shows a barcode rank plot with a characteristic "cliff-and-knee" shape, indicating good separation between cells and background [82].
Manual Filtering with Loupe Browser
For interactive exploration and filtering, open the .cloupe file in Loupe Browser to:
- Visualize the distribution of UMI counts, number of features, and mitochondrial percentages
- Apply filters using the slider interfaces to remove outliers
- Document all filtering thresholds for reproducibility [82]
Advanced QC Considerations
For specialized applications, consider these additional approaches:
- Ambient RNA Removal: Use tools like SoupX or CellBender to correct for background noise caused by free-floating RNA from lysed cells, particularly important when studying rare cell types [82].
- Doublet Detection: Employ algorithms like Scrublet or DoubletFinder to identify and remove multiplets, especially crucial in high-throughput experiments where doublet rates increase with cell loading concentrations.
Phase 2: Normalization
Theoretical Foundation
Normalization addresses technical variations in scRNA-seq data to ensure that gene expression comparisons are biologically meaningful rather than reflecting technical artifacts. The primary sources of technical variation include sequencing depth (library size), capture efficiency, amplification biases, and gene length effects [70]. Unlike bulk RNA-seq, scRNA-seq data exhibits unique characteristics such as zero inflation (excess of zero counts) and high variability that require specialized normalization approaches. The core objective is to remove technical biases while preserving true biological heterogeneity, enabling valid comparisons of gene expression patterns across cells.
Normalization Techniques and Applications
Various normalization strategies have been developed for scRNA-seq data, each with distinct assumptions and applications.
Table 2: scRNA-seq Normalization Methods and Their Applications
| Method Category | Key Principles | Advantages | Limitations | Common Tools |
|---|---|---|---|---|
| Library Size Normalization | Scales counts by total UMI counts per cell (e.g., CPM, TPM) | Simple, intuitive, computationally efficient | Does not address sequence composition effects | Seurat, Scanpy [81] |
| Global Scaling Methods | Uses a size factor derived from pool-based cells to normalize | Robust to outliers, handles composition biases | Assumes most genes are not differentially expressed | SCTransform, scran [70] |
| Downsampling Methods | Randomly subsamples counts to equal levels across cells | Avoids distributional assumptions, preserves relationships | Discards data, may increase technical noise | DCA, scVI [70] |
Practical Protocol: Standard Normalization Workflow
Log-Normalization with Seurat
A widely used approach implemented in Seurat involves:
- Total Count Normalization: Normalize the raw count data for each cell by its total UMI count, multiplying by a scale factor (e.g., 10,000), thus generating transcripts per 10,000 (TP10K).
- Log Transformation: Apply a natural log transformation using
log1p(log(1+x)) to the normalized values to dampen the effect of extreme outliers. - Feature Selection: Identify highly variable genes (HVGs) that exhibit high cell-to-cell variation, typically selecting 1,000-5,000 genes for downstream analyses. For optimal performance, scEVE selects 1,000 highly variable genes using the
FindVariableFeatures()function from Seurat [81].
This normalized data then serves as input for dimensionality reduction techniques like PCA, which is a prerequisite for clustering analyses.
Advanced Normalization Considerations
For complex experimental designs, additional normalization strategies may be required:
- Batch Effect Correction: When integrating multiple datasets or samples, use integration methods like Harmony, CCA (in Seurat), or BBKNN to remove technical variations between batches while preserving biological variance.
- Depth Normalization: Address variations in sequencing depth using deconvolution methods implemented in the scran package, which pools cells with similar expression profiles to estimate size factors more accurately.
Phase 3: Clustering
Conceptual Framework
Clustering analysis groups cells based on the similarity of their transcriptomic profiles, enabling the identification of distinct cell types, states, and subpopulations within seemingly homogeneous tissues [21]. This represents a fundamental step in scRNA-seq analysis, as nearly all downstream biological interpretations—from differential expression to trajectory inference—depend on accurate cluster definitions. The core challenge lies in distinguishing true biological signals from technical noise while determining the appropriate resolution at which to analyze cellular heterogeneity. Remarkably, as of January 2024, over 375 clustering methods applicable to scRNA-seq datasets have been documented, each employing distinct algorithmic approaches and hypotheses [81].
Clustering Algorithms and Ensemble Methods
The table below summarizes major clustering approaches used in scRNA-seq analysis, along with state-of-the-art ensemble methods that integrate multiple algorithms.
Table 3: scRNA-seq Clustering Methods and Performance Characteristics
| Method Category | Representative Algorithms | Key Principles | Performance Notes |
|---|---|---|---|
| Community Detection | Monocle3, Seurat | Identifies densely connected node groups in graphs | Monocle3 ranked 1/14, Seurat 5/14 in benchmark [81] |
| Stability Metrics | densityCut | Uses diffusion-based distance and stability metrics | Ranked 13/14 in benchmark by Yu et al. [81] |
| Inter/Intra-cluster Similarity | SHARP | Leverages intercellular and intracellular similarities | Ranked 14/14 in benchmark by Yu et al. [81] |
| Ensemble Methods | scEVE | Integrates multiple methods to generate consensus | Outperforms individual methods; provides uncertainty values [81] |
Practical Protocol: Standard Clustering Pipeline
Dimensionality Reduction and Graph-Based Clustering
A standard clustering workflow in Seurat includes:
- Principal Component Analysis (PCA): Perform linear dimensionality reduction on the normalized and scaled data, selecting significant PCs based on the elbow plot of standard deviations.
- Graph Construction: Build a k-nearest neighbor (KNN) graph in PCA space, typically using the first 10-30 principal components.
- Community Detection: Apply modularity optimization algorithms (e.g., Louvain, Leiden) to partition the graph into clusters, which represent putative cell populations.
- Resolution Parameter Tuning: Adjust the resolution parameter to control the granularity of clustering, with higher values resulting in more clusters.
Ensemble Clustering with scEVE
For enhanced robustness, consider implementing ensemble approaches like scEVE, which addresses methodological bias by integrating multiple clustering methods:
- Base Cluster Generation: Apply multiple computationally efficient and methodologically diverse clustering methods (e.g., monocle3, Seurat, densityCut, SHARP) to the same dataset [81].
- Pairwise Similarity Calculation: Compute minimal proportion of cells shared between base clusters using the formula: [ S{x,y} = \min\left(\frac{N{x \cap y}}{Nx}, \frac{N{x \cap y}}{Ny}\right) ] where ( S{x,y} ) is the similarity between base clusters x and y, ( N{x \cap y} ) is the number of cells in both clusters, and ( Nx ) is the number of cells in cluster x [81].
- Robust Cluster Identification: Identify clusters that consistently appear across multiple methods by detecting strong pairwise similarities (typically with threshold ( S_{lim} = 0.5 )) between base clusters [81].
- Robustness Quantification: scEVE provides uncertainty values for clustering results and enables multi-resolution analysis, addressing two grand challenges in single-cell data science [81].
Cluster Annotation and Validation
Following cluster identification, validate and interpret results through:
- Differential Expression Analysis: Identify marker genes that are significantly enriched in each cluster compared to all others.
- Cell Type Annotation: Map clusters to known cell types using marker databases and reference datasets.
- Biological Validation: Confirm cluster identities through known marker expression and functional enrichment analyses.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of scRNA-seq bioinformatics pipelines requires both computational tools and wet-lab reagents. The following table outlines key solutions and their functions in the experimental workflow.
Table 4: Essential Research Reagent Solutions for scRNA-seq Workflows
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| Chromium GEM Kits (10x Genomics) | Partition cells into Gel Bead-in-Emulsions (GEMs) for barcoding | Universal assays for 3' or 5' gene expression; Flex assays enable frozen/fixed sample processing [12] |
| Cell Barcoding Oligonucleotides | Uniquely label mRNA from individual cells during reverse transcription | Contains poly(dT) primers, cell barcode, and UMI; critical for single-cell resolution [70] [12] |
| Single Cell Multiplexing Reagents | Pool multiple samples by labeling with sample-specific barcodes | Reduces batch effects and costs; enables larger experimental designs |
| Viability Stains | Distinguish live from dead cells during sample preparation | Critical for ensuring high-quality input material; reduces ambient RNA background |
Quality control, normalization, and clustering represent three interconnected pillars of scRNA-seq bioinformatics pipelines, each playing an indispensable role in transforming raw sequencing data into biologically meaningful insights. As the field continues to evolve with emerging technologies like spatial transcriptomics and multi-omics integration, these foundational steps will remain essential for ensuring data quality and interpretability. The integration of ensemble methods like scEVE demonstrates how the field is advancing toward more robust, reproducible analyses that quantify uncertainty and enable multi-resolution exploration of cellular heterogeneity [81]. For researchers embarking on scRNA-seq studies, adherence to these standardized protocols provides a solid foundation for uncovering the complex cellular dynamics that underlie development, disease, and therapeutic responses, ultimately advancing the development of precision medicine approaches.
The advent of single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity in complex tissues. However, transcriptomics alone provides only a one-dimensional view of the complex regulatory machinery governing cellular identity and function [83]. The integration of scRNA-seq with other molecular modalities, such as single-cell ATAC-seq (scATAC-seq) for chromatin accessibility and proteomic assays for surface protein abundance, enables a more comprehensive dissection of cellular states and regulatory networks [84] [85]. This multi-omics approach has become increasingly vital for uncovering the intricate relationships between different layers of molecular regulation that drive cellular differentiation, immune responses, and disease pathogenesis [86] [83].
The fundamental challenge in single-cell multi-omics lies in the effective computational integration of datasets with distinct feature spaces and statistical characteristics. While scRNA-seq measures the expression levels of thousands of genes across individual cells, scATAC-seq captures chromatin accessibility patterns across the genome, and proteomic assays quantify the abundance of specific proteins [84] [85]. Successful integration requires methods that can align these disparate data types into a unified analytical framework while preserving biological meaningful variation and removing technical artifacts [86]. The field has responded with a rapid proliferation of computational tools, making method selection and implementation a critical consideration for researchers embarking on multi-omics studies [84].
Computational Strategies for Data Integration
Categories of Integration Methods
Computational methods for integrating single-cell multi-omics data can be broadly classified into three main categories based on the nature of the data they are designed to handle [84]. Unpaired integration methods are designed for datasets where different omics modalities are profiled in different cells from the same tissue or biological system. These methods include LIGER, which performs integrative non-negative matrix factorization (iNMF) to learn shared metagenes [84]; Seurat v3, which combines canonical correlation analysis (CCA) with mutual nearest neighbors (MNNs) to identify integration anchors [84]; and GLUE, which incorporates knowledge-based graphs with adversarial multimodal alignment [84]. Paired integration methods handle data where multiple omics are simultaneously profiled from the same individual cells. This category includes MOFA+, which uses variational inference to gain a low-dimensional representation across different omics [84], and scMVP, which employs a clustering-constrained multi-view variational autoencoder [84]. Paired-guided integration methods leverage a small set of paired multi-omics data to guide the integration of larger unpaired datasets, with MultiVI and Cobolt both utilizing deep generative models for this purpose [84].
Benchmarking Integration Performance
Recent benchmarking studies have evaluated integration methods across multiple performance aspects. A comprehensive assessment of 12 popular methods considered six key evaluation metrics: neighborhood overlap score (NOS) for omics mixing, graph connectivity (GC), Seurat alignment score (SAS), average silhouette width across omics (ASW-O) for batch mixing, and mean average precision (MAP) and normalized mutual information (NMI) for cell type conservation [84]. Performance varies across methods, with some excelling in specific tasks. For instance, Harmony demonstrates strong performance for simpler integration tasks with distinct batch structures, while deep-learning approaches like scANVI and scVI perform better for complex atlas-level integration [86]. The runtime and memory usage of these methods also differ significantly, with methods like SnapATAC2 demonstrating linear scalability and substantially reduced memory requirements compared to traditional approaches [87].
Table 1: Performance Comparison of Selected Multi-Omics Integration Methods
| Method | Category | Key Algorithm | Strengths | Scalability |
|---|---|---|---|---|
| Harmony | Unpaired | Soft k-means clustering | Effective for simpler batch correction [86] | Linear runtime [86] |
| LIGER | Unpaired | Integrative NMF | Identifies shared and modality-specific factors [84] | Moderate [84] |
| Seurat v3 | Unpaired | CCA + MNNs | Well-established, comprehensive toolkit [84] | Moderate [84] |
| GLUE | Unpaired | Graph-based + adversarial alignment | Incorporates prior knowledge [84] | High with GPU [84] |
| MOFA+ | Paired | Variational inference | Handles multiple omics modalities [84] | Moderate [84] |
| scMVP | Paired | Variational autoencoder | Clustering consistency constraints [84] | High with GPU [84] |
| MultiVI | Paired-guided | Deep generative model | Leverages paired data as guide [84] | High with GPU [84] |
| scMODAL | Unpaired | Neural networks + GANs | Works with limited linked features [85] | High with GPU [85] |
Experimental Design and Workflow
Sample Preparation and Single-Cell Sequencing
The foundation of any successful multi-omics study begins with proper sample preparation and single-cell isolation. The process typically starts with generating a high-quality single-cell suspension from the tissue of interest through enzymatic digestion and/or mechanical dissociation [88]. It is crucial to optimize this step to maximize cell viability and minimize stress-induced alterations to the transcriptome or epigenome. Cells are then isolated using various technologies, including fluorescence-activated cell sorting (FACS), microfluidic systems, or droplet-based encapsulation [88]. For scRNA-seq, isolated cells undergo cell lysis, followed by mRNA capture using poly(T) primers, reverse transcription to complementary DNA (cDNA), and amplification via PCR or in vitro transcription [88]. Unique molecular identifiers (UMIs) and cell barcodes are incorporated during these steps to enable accurate quantification and distinguish individual cells [88].
For scATAC-seq, the protocol centers on using the Tn5 transposase to tag open chromatin regions with sequencing adaptors [84]. The transposase simultaneously fragments the DNA and adds adaptor sequences in accessible regions, which are then amplified and sequenced [84]. When integrating scRNA-seq with proteomic data through CITE-seq, antibody-derived tags (ADTs) with DNA barcodes are used to label surface proteins, allowing simultaneous quantification of transcriptome and proteome in the same cell [85]. The compatibility of sample preparation across modalities must be carefully considered when planning multi-omics studies.
Data Preprocessing and Quality Control
Robust preprocessing and quality control (QC) are essential for generating reliable multi-omics data. The initial processing of sequencing data involves demultiplexing raw FASTQ files, read alignment to the appropriate reference genome (using tools like STAR for scRNA-seq), and quantification of features (genes, chromatin peaks, or proteins) per cell [89]. For scRNA-seq data, QC metrics should include assessments of UMI counts per cell, genes detected per cell, and mitochondrial gene percentage to identify low-quality cells or empty droplets [86] [89]. Similarly, for scATAC-seq data, metrics such as fraction of fragments in peaks and nucleosome banding pattern should be evaluated [90].
Ambient RNA contamination and doublets pose significant challenges in single-cell data. Methods like SoupX and CellBender can estimate and remove ambient RNA contamination [86], while tools like scDblFinder have demonstrated high accuracy in detecting doublets [86]. Normalization strategies must be carefully selected based on the data characteristics and downstream analysis goals. The shifted logarithm transformation performs well for variance stabilization prior to dimensionality reduction, while Scran normalization using pool-based size factors is effective for batch correction tasks [86]. For scATAC-seq data, the question of whether to treat measurements as qualitative or quantitative remains debated, with different implications for downstream differential accessibility analysis [90].
Diagram 1: Integrated Workflow for Single-Cell Multi-Omics Analysis. This workflow encompasses both experimental and computational phases, highlighting key steps from sample preparation to biological interpretation.
Practical Implementation Protocol
Step-by-Step Integration Procedure
This protocol provides a detailed guide for integrating scRNA-seq with scATAC-seq and proteomic data using the Seurat framework, which has demonstrated robust performance across various integration tasks [84] [86]. Before beginning, ensure all necessary software packages are installed: Seurat, Signac for scATAC-seq data, and any modality-specific packages.
Step 1: Data Input and Initial Processing Load the scRNA-seq count matrix and create a Seurat object. Perform standard preprocessing including normalization (SCTransform recommended), feature selection, and scaling. For scATAC-seq data, create a Seurat object using the peak-by-cell matrix and perform term frequency-inverse document frequency (TF-IDF) normalization [86]. For proteomic data (e.g., CITE-seq ADT counts), include the ADT matrix as an additional assay in the Seurat object.
Step 2: Modality-Specific Dimension Reduction For scRNA-seq data, run principal component analysis (PCA) on the variable genes. For scATAC-seq data, perform latent semantic indexing (LSI) on the TF-IDF matrix [86]. These modality-specific reductions will serve as input for integration.
Step 3: Identification of Integration Anchors Using the FindTransferAnchors function in Seurat, identify cross-modality anchors between the scRNA-seq and scATAC-seq datasets. The accuracy of this step can be improved by using a reference-based approach if well-annotated scRNA-seq data is available [86]. For proteomic integration, consider using methods like bindSC or MaxFuse that are specifically designed for modalities with potentially weak feature relationships [85].
Step 4: Data Transfer and Integration Transfer data across modalities using the TransferData function. For scATAC-seq to scRNA-seq integration, this typically involves imputing gene expression based on chromatin accessibility [86]. Alternatively, compute a gene activity matrix from scATAC-seq data by summing accessibility peaks near each gene's promoter and gene body [84]. For proteomic data, protein abundances can be directly integrated with transcriptional data using CCA-based methods [85].
Step 5: Joint Visualization and Analysis Merge the datasets and perform joint dimension reduction using UMAP based on the integrated features. The success of integration should be evaluated using both qualitative assessment of visualization and quantitative metrics such as local structure preservation and cell type conservation [84].
Quality Assessment and Troubleshooting
After integration, systematically assess the quality using multiple approaches. Visual inspection of UMAP plots should show mixing of cells from different modalities within the same cell type while maintaining separation between distinct cell types [84]. Quantitatively, calculate the average silhouette width across omics (ASW-O) to assess mixing and normalized mutual information (NMI) to evaluate cell type conservation [84].
Common integration issues include over-correction (where biological variation is removed along with technical effects) and under-correction (where batch effects persist). If over-correction occurs, reduce the strength of the integration parameters. If under-correction is observed, increase the dimensionality parameter in FindTransferAnchors or consider using a different integration method such as Harmony or scMODAL [84] [85]. For particularly challenging integrations with limited shared features, scMODAL has demonstrated robust performance even with very few linked features by leveraging neural networks and generative adversarial networks [85].
Table 2: Essential Research Reagent Solutions for Single-Cell Multi-Omics
| Reagent/Resource | Function | Example Applications |
|---|---|---|
| 10x Genomics Chromium | Droplet-based single-cell partitioning | scRNA-seq, scATAC-seq, multiome (RNA+ATAC) [2] |
| BD Rhapsody | Microwell-based single-cell capture | scRNA-seq with protein detection (AbSeq) [89] |
| CITE-seq Antibodies | Oligo-tagged antibodies for protein detection | Simultaneous transcriptome and proteome profiling [85] |
| Tn5 Transposase | Tagmentation of accessible chromatin | scATAC-seq library preparation [84] |
| UMI Barcodes | Unique molecular identifiers for quantification | Distinguishing biological signals from amplification noise [88] |
| Cell Hashtags | Sample multiplexing with lipid-tagged oligos | Pooling multiple samples while retaining sample identity [83] |
Downstream Analysis and Biological Interpretation
Advanced Analytical Applications
Once successfully integrated, multi-omics data enables sophisticated analytical approaches that reveal deeper biological insights. Regulatory network inference can connect transcription factors identified through scATAC-seq with their target genes measured in scRNA-seq [84]. Tools like SCENIC can be applied to infer gene regulatory networks from integrated data [83]. Trajectory analysis using methods like Monocle3 or RNA velocity can reconstruct developmental pathways while incorporating epigenetic dynamics [89] [83]. For studying cellular communication, cell-cell communication inference methods can be enhanced by incorporating surface protein expression from proteomic data to improve ligand-receptor interaction predictions [83].
Differential analysis in multi-omics contexts requires special consideration. For scATAC-seq data specifically, benchmarking studies have shown that pseudobulk methods generally outperform approaches designed for single-cell RNA-seq when identifying differentially accessible regions [90]. The Wilcoxon rank-sum test is the most widely used method in published scATAC-seq analyses, though consistency in analytical approaches remains limited across the field [90]. When analyzing multi-omics data, it is crucial to perform differential analysis across all modalities simultaneously to identify coordinated changes in gene expression, chromatin accessibility, and protein abundance.
Visualization and Result Interpretation
Effective visualization is essential for interpreting multi-omics results. UMAP plots should be generated showing both modality mixing and cell type annotations [84]. Heatmaps can display matched patterns of gene expression and chromatin accessibility across cell types [85]. For visualizing regulatory connections, circos plots or network diagrams can illustrate relationships between distal regulatory elements and their potential target genes [84].
Biological validation of findings is crucial. For identified regulatory elements, motif enrichment analysis can confirm the presence of expected transcription factor binding sites [87]. Candidate biomarkers derived from integrated analysis should be validated using orthogonal methods such as fluorescence in situ hybridization or immunohistochemistry [83]. When interpreting results, consider the limitations of each technology—scATAC-seq may miss some relevant regulatory elements due to sparse coverage, while proteomic data may not reflect intracellular protein dynamics [85].
The integration of scRNA-seq with ATAC-seq and proteomic data represents a powerful approach for comprehensive cellular characterization. As benchmarking studies have shown, method selection should be guided by specific research questions and data characteristics [84]. While current methods have enabled robust integration, challenges remain in handling extremely large datasets, integrating modalities with weak feature relationships, and accurately inferring causal regulatory relationships [85].
Emerging technologies and computational approaches promise to further advance single-cell multi-omics. The development of deep learning frameworks like scMODAL that can effectively integrate modalities with limited known feature relationships addresses a critical challenge in the field [85]. Improvements in spatial multi-omics will enable the incorporation of spatial context into integrated analyses [83]. As the scale of single-cell studies continues to grow, computational efficiency will remain a key consideration, with tools like SnapATAC2 demonstrating the importance of scalable algorithms for large-scale data [87].
For researchers embarking on single-cell multi-omics studies, following best practices in experimental design, quality control, and method selection will maximize the insights gained from these powerful approaches. By effectively integrating multiple molecular modalities, we can move beyond transcriptomics to develop unified models of cellular function and regulation across molecular layers.
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity by profiling gene expression at individual cell resolution. However, a significant limitation of standard scRNA-seq protocols is the requirement for tissue dissociation, which completely destroys the native spatial organization of cells within their tissue microenvironment [91]. This spatial context is not merely structural—it fundamentally regulates cellular function through localized gene expression patterns, cell-cell communication, and tissue-scale gradients of signaling molecules.
Spatial validation bridges this critical gap by confirming the precise localization of cell types and gene expression patterns identified through scRNA-seq. By preserving architectural relationships, spatial transcriptomics technologies enable researchers to answer fundamental biological questions about cellular neighborhoods, pathogenic mechanisms in disease contexts, and the functional organization of complex tissues. This application note provides a structured framework for employing RNA fluorescence in situ hybridization (FISH) and spatial transcriptomics to validate findings from single-cell RNA sequencing projects.
Platform Selection: Matching Technology to Validation Goals
Choosing the appropriate spatial transcriptomics platform requires careful consideration of resolution, multiplexing capability, and sample compatibility. The table below summarizes key performance characteristics of major imaging-based spatial transcriptomics (iST) platforms based on recent benchmarking studies.
Table 1: Benchmarking Comparison of Commercial Imaging Spatial Transcriptomics Platforms for FFPE Tissues
| Platform | Spatial Resolution | Transcript Detection Sensitivity | Key Strengths | Optimal Use Cases |
|---|---|---|---|---|
| 10X Xenium | Subcellular | Higher transcript counts per gene without sacrificing specificity [92] | High sensitivity, low false discovery rates in cell typing [92] | Validating cell type localization and detecting low-abundance transcripts |
| Vizgen MERSCOPE | Subcellular | Moderate sensitivity | Direct probe hybridization with extensive tiling [92] | Whole-transcriptome spatial mapping with error-robust encoding |
| Nanostring CosMx | Subcellular | High transcript counts, concordance with scRNA-seq [92] | Ability to resolve slightly more cell clusters [92] | High-plex validation in complex tissue microenvironments |
For specialized applications requiring live-cell analysis, emerging technologies now enable multiplexed RNA imaging in living cells. These approaches utilize synthetic oligonucleotide probes, CRISPR-dCas systems, and fluorogenic RNA aptamers to monitor RNA dynamics in real time, providing temporal resolution that fixed-cell methods cannot capture [93].
Integrated Workflow for Spatial Validation
The following diagram illustrates the comprehensive workflow for spatial validation, from single-cell analysis through spatial confirmation and data integration:
Diagram 1: Spatial validation workflow from scRNA-seq to biological confirmation.
Critical Pre-Experimental Considerations
Before initiating wet-lab work, several foundational decisions determine validation success:
Research Question Alignment: Confirm that spatial resolution is essential for your biological question. Spatial validation excels when investigating cell-cell interactions, tissue architecture, or microenvironmental gradients, but may be unnecessary for global transcriptional comparisons [94].
Team Assembly: Spatial technologies require multidisciplinary expertise. Ensure involvement of wet-lab specialists, pathologists for tissue annotation, and bioinformaticians for data analysis from the project's inception [94].
Experimental Replication: Account for spatial heterogeneity through appropriate biological replicates. Underpowered studies represent a common pitfall in spatial validation; consult statistical resources or experienced cores during planning phases [94].
Experimental Protocols for Spatial Validation
Sample Preparation and Quality Control
Proper tissue processing is paramount for successful spatial validation:
Preservation Strategy Selection: Fresh-frozen tissue generally provides higher RNA integrity, while Formalin-Fixed Paraffin-Embedded (FFPE) tissues enable access to vast archival biobanks but may exhibit reduced RNA quality [92] [94].
Section Quality Control: Optimize section thickness based on platform requirements (typically 5-10 μm). Consecutive sections should be collected for H&E staining and spatial analysis to enable pathological annotation [94].
RNA Integrity Assessment: For FFPE samples, determine RNA integrity numbers (RIN) or DV200 values (>60% recommended for MERSCOPE). Implement rigorous sample screening procedures to exclude degraded samples [92].
Probe and Panel Design
Effective probe design significantly impacts detection sensitivity and specificity:
Computational Design Tools: Platforms like TrueProbes utilize genome-wide BLAST-based binding analysis with thermodynamic modeling to generate high-specificity probe sets, outperforming alternative tools in both computational metrics and experimental validation [95].
Panel Optimization: For targeted validation, include positive control genes with known expression patterns and negative controls to assess background signal. When using commercial panels, select genes that definitively distinguish cell populations identified in scRNA-seq [94].
Table 2: Essential Research Reagents and Computational Tools for Spatial Validation
| Category | Specific Tool/Reagent | Function | Application Context |
|---|---|---|---|
| Probe Design | TrueProbes | Designs high-specificity FISH probes using thermodynamic modeling | Optimizing signal-to-noise ratio for custom target validation |
| Cell Segmentation | Proseg | Defines cell boundaries using probabilistic modeling of RNA distribution | Improving accuracy of single-cell resolution in dense tissues |
| Data Processing | PIPEFISH | Processes raw FISH images into spatially annotated transcripts | Standardizing analysis across MERFISH, seqFISH, and ISS platforms |
| Image Analysis | QuantISH | Quantifies RNA expression in chromogenic or fluorescent ISH images | Automated analysis of CISH images without multiple channels |
| Commercial Platforms | Xenium, MERSCOPE, CosMx | Integrated hardware/software for spatial transcriptomics | Targeted validation studies with optimized workflows |
Wet-Lab Execution
Spatial transcriptomics protocols are often rigid and require meticulous execution:
Pre-Experiment Setup: Ensure reagents are fresh, properly stored, and equilibrated to correct temperatures. Prepare dedicated workspace to prevent RNAase contamination [94].
Hybridization Optimization: For multiplexed FISH methods, systematically optimize hybridization conditions including probe concentration, hybridization duration, and buffer composition. Recent empirical studies indicate that modifications to these parameters can dramatically improve signal quality [96].
Image Acquisition Standardization: Establish consistent imaging parameters across all samples. Include control fields for exposure calibration and monitor signal bleaching across multiple rounds of imaging [96].
Data Processing and Analysis Framework
Cell Segmentation Strategies
Accurate cell segmentation remains a critical challenge in spatial transcriptomics. Traditional segmentation methods based on antibody staining frequently misidentify cellular borders, leading to incorrect assignment of transcripts [91]. Advanced computational tools like Proseg address this limitation by using probabilistic models that define cell boundaries based on RNA distribution patterns, effectively treating cells as "bags of randomly distributed RNA" [91]. This approach has been shown to significantly reduce suspicious gene co-expression events resulting from segmentation errors.
Analytical Quality Control
Implement rigorous quality control metrics throughout the analytical pipeline:
Internal QC Metrics: Utilize platform-specific quality indicators such as transcripts per cell, background fluorescence, and positive control gene expression [97].
Segmentation Validation: Manually review segmentation boundaries across diverse tissue regions, particularly in areas with high cell density or unusual morphology [91].
Concordance Assessment: Compare spatial results with original scRNA-seq findings, identifying both confirming and discordant patterns for biological interpretation.
The following diagram illustrates the core computational workflow for processing spatial transcriptomics data:
Diagram 2: Core computational workflow for spatial transcriptomics data processing.
Integration with Single-Cell RNA Sequencing Data
Effective spatial validation requires sophisticated integration of scRNA-seq and spatial data:
Cell Type Mapping: Transfer cell type labels from scRNA-seq clusters to spatially resolved cells using integration algorithms that leverage common gene expression features [94].
Spatial Neighborhood Analysis: Identify recurrent cellular neighborhoods and interaction patterns that may drive the biological phenomena under investigation.
Validation Metrics: Quantitatively assess concordance between scRNA-seq predictions and spatial observations, reporting both confirmation rates and discovered discrepancies.
Troubleshooting and Optimization Guidelines
Common challenges in spatial validation experiments include:
Low Signal-to-Noise Ratio: Optimize probe design using tools like TrueProbes, increase probe concentration within optimal ranges, and extend hybridization time while maintaining specificity [95].
Cell Segmentation Failures: Implement advanced segmentation tools like Proseg that use RNA distribution rather than only nuclear staining, particularly beneficial in dense tissues or tumors [91].
Spatial Resolution Limitations: For technologies with lower inherent resolution, prioritize validation of distinctly localized cell types rather than subtle spatial gradients.
Future Perspectives and Emerging Technologies
The spatial transcriptomics field continues to evolve rapidly, with several promising directions:
Multi-Omic Integration: Combining spatial transcriptomics with proteomics, epigenomics, and metabolomics provides richer contextual understanding of cellular states [94].
Live-Cell Imaging: New fluorescent probes now enable multiplexed RNA imaging in living cells, opening possibilities for tracking RNA dynamics and interactions in real time [93].
Computational Advancements: Improved algorithms for cell segmentation, data integration, and spatial pattern recognition continue to enhance the resolution and accuracy of spatial validation [91] [97].
As these technologies mature, spatial validation will evolve from a confirmatory method to a discovery tool that generates novel hypotheses about tissue organization and function in development, homeostasis, and disease.
Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to characterize cellular heterogeneity within complex tissues by profiling the transcriptome of individual cells. However, a significant limitation of this powerful technique is the destruction of native spatial context during tissue dissociation, which eliminates crucial information about protein localization and cellular microenvironment. Furthermore, mRNA expression levels do not always correlate directly with functional protein abundance due to post-transcriptional regulation and protein turnover dynamics. This disconnect underscores the essential need for protein-level validation techniques to corroborate scRNA-seq findings.
Immunofluorescence (IF) and Immunohistochemistry (IHC) serve as cornerstone methodologies for protein validation, allowing researchers to visualize target protein expression within preserved tissue architecture. When strategically integrated with scRNA-seq data, these techniques provide a comprehensive understanding of gene expression at both transcriptional and translational levels, while maintaining critical spatial information lost in single-cell isolation procedures. This application note details how IF and IHC can be systematically employed to verify and contextualize scRNA-seq discoveries, with particular emphasis on formalin-fixed paraffin-embedded (FFPE) tissue protocols that enhance sample compatibility across methodologies.
Technical Comparison: IHC vs. IF for scRNA-seq Corroboration
Fundamental Principles and Detection Mechanisms
Immunohistochemistry (IHC) utilizes antibodies conjugated to enzymes such as horseradish peroxidase (HRP) or alkaline phosphatase (AP) that catalyze chromogenic reactions (e.g., DAB, AEC) to produce visible color precipitates at antigen sites. This permanent staining is compatible with standard brightfield microscopy and provides excellent morphological context preferred by pathologists for diagnostic workflows [98] [99].
Immunofluorescence (IF) employs fluorescent dye-conjugated antibodies that emit light of specific wavelengths when excited by appropriate light sources. The resulting signals are detected using fluorescence microscopy, enabling superior sensitivity and the capacity for multiplexing multiple protein targets simultaneously through spectral discrimination [98] [100].
Comparative Analysis for Research Applications
Table 1: Comprehensive Comparison of IHC and IF for Protein Validation
| Parameter | Immunohistochemistry (IHC) | Immunofluorescence (IF) | Relevance to scRNA-seq Integration |
|---|---|---|---|
| Detection Chemistry | Chromogenic enzymes (HRP/AP + DAB) | Fluorophores (Alexa Fluor, Cy dyes) | IF enables multiplexing for co-localization of proteins corresponding to scRNA-seq identified markers |
| Max Markers/Slide | 1-2 markers typically | 2-8 markers (standard); Up to 60 with ultra-high-plex platforms [98] | High-plex IF aligns with multi-gene expression patterns from scRNA-seq clusters |
| Spatial Context | Preserves tissue architecture | Preserves tissue architecture with subcellular resolution | Both maintain spatial context lost in scRNA-seq processing |
| Signal Stability | Permanent, archivable slides | Moderate (photobleaching risk) | IHC preferable for long-term validation studies |
| Sensitivity | Moderate | High to very high | IF better for low-abundance proteins potentially missed in scRNA-seq |
| Equipment | Brightfield microscope | Fluorescence microscope + scanner | IF requires more specialized equipment but provides higher resolution data |
| Morphology Quality | Excellent for pathological assessment | Good to moderate | IHC preferred when correlating protein expression with histological features |
| Typical Turnaround | 3-5 days [98] | 5-7 days (standard); 7-10 days (high-plex) [98] | Planning consideration for integrated study timelines |
| Compatibility with RNA-seq | FFPE samples compatible with spatial transcriptomics | FFPE samples compatible with emerging spatial technologies [100] [101] | Both techniques compatible with adjacent sections for spatial correlation |
Table 2: Decision Framework for Technique Selection
| Research Scenario | Recommended Technique | Rationale |
|---|---|---|
| Validation of 1-2 key protein targets | IHC | Simplified workflow, permanent record, cost-effective |
| Multiplex protein detection (3+ markers) | IF | Simultaneous visualization of multiple proteins from related scRNA-seq pathways |
| Subcellular localization studies | IF | Superior resolution for cytoplasmic/nuclear/membrane demarcation |
| Archival tissue studies | IHC (FFPE-compatible) | Long-term stability, compatible with clinical samples |
| Live cell imaging | IF (on cultured cells) | Compatibility with live-cell applications |
| Spatial transcriptomics correlation | Either (FFPE-compatible) | Direct comparison with adjacent tissue sections |
| High-throughput screening | IF with automated scanning | Rapid quantification potential |
| Clinical diagnostic correlation | IHC | Standard for pathology, facilitates translational interpretation |
Integrated Experimental Protocols
Multiplex Immunofluorescence for FFPE Tissues
This protocol adapts IF for formalin-fixed paraffin-embedded (FFPE) tissues, enhancing compatibility with scRNA-seq validation studies by allowing investigation of archived samples [100].
Materials and Reagents
- Primary Antibodies: Validate species compatibility for multiplexing (e.g., mouse IgG1, mouse IgG2b, mouse IgM, rabbit IgG)
- Secondary Antibodies: Species-specific conjugates with minimal cross-reactivity (e.g., goat anti-mouse IgG1 Alexa Fluor 488, goat anti-mouse IgG2b Alexa Fluor 647, goat anti-mouse IgM Alexa Fluor 594)
- Blocking Buffer: 5% normal serum from secondary antibody host species in PBS
- Antigen Retrieval Buffer: Tris-EDTA, pH 9.0 or citrate-based, pH 6.0
- Mounting Medium: Anti-fade mounting medium with DAPI for nuclear counterstaining
Methodology
- Tissue Sectioning: Cut FFPE sections at 4-5 μm thickness using a microtome and mount on charged slides.
- Deparaffinization: Immerse slides in xylene (3 changes, 5 minutes each) followed by rehydration through graded ethanol series (100%, 95%, 70%) to distilled water.
- Antigen Retrieval: Heat-induced epitope retrieval using appropriate buffer in a decloaking chamber or water bath (95-100°C) for 20 minutes, followed by 20-minute cool-down at room temperature.
- Permeabilization: Incubate with PBS containing 0.25% Triton X-100 for 10 minutes.
- Blocking: Apply blocking buffer for 1 hour at room temperature in a humidified chamber.
- Primary Antibody Incubation: Apply optimized antibody cocktails diluted in blocking buffer overnight at 4°C in a humidified chamber.
- Washing: Rinse slides with PBS containing 0.025% Triton X-100 (3 × 5 minutes).
- Secondary Antibody Incubation: Apply fluorophore-conjugated secondary antibodies diluted in blocking buffer for 1 hour at room temperature protected from light.
- Counterstaining and Mounting: Apply DAPI (1 μg/mL) for 5 minutes, rinse with distilled water, and mount with anti-fade medium.
- Imaging: Acquire images using a fluorescence microscope or high-content imaging system with appropriate filter sets for each fluorophore.
Immunohistochemistry for Sequential Validation
For validating individual protein targets identified through scRNA-seq analysis, this IHC protocol provides robust, quantitative results.
Materials and Reagents
- Primary Antibodies: Optimized for IHC with known specificity
- Detection System: HRP-based detection kit with DAB chromogen
- Hematoxylin: For nuclear counterstaining
- Blocking Solution: Serum-free protein block with endogenous enzyme blocking
Methodology
- Section Preparation: Follow steps 1-3 from the IF protocol above.
- Endogenous Peroxidase Blocking: Incubate with 3% hydrogen peroxide for 10 minutes to quench endogenous peroxidase activity.
- Protein Blocking: Apply protein block for 10 minutes to reduce non-specific binding.
- Primary Antibody Incubation: Apply optimized primary antibody dilution for 1 hour at room temperature or overnight at 4°C.
- Detection: Incubate with HRP-conjugated secondary antibody or polymer-based detection system for 30 minutes, followed by DAB chromogen application (typically 5-10 minutes).
- Counterstaining: Immerse in hematoxylin for 30-60 seconds, followed by blueing in tap water.
- Dehydration and Mounting: Dehydrate through graded alcohols, clear in xylene, and mount with permanent mounting medium.
- Image Analysis: Digitize slides using a whole-slide scanner and quantify staining intensity using digital pathology software.
Integration with Single-Cell RNA Sequencing Data
Strategic Corroboration Approach
The integration of IF/IHC with scRNA-seq data requires a systematic validation strategy to maximize confirmatory value:
Target Selection: Prioritize protein targets based on scRNA-seq analysis:
- Differentially expressed genes between cell subpopulations
- Key marker genes defining novel cell clusters
- Genes encoding surface receptors or secreted proteins implicated in cell-cell communication
- Transcription factors driving cellular identity
Spatial Contextualization: Localize protein expression within tissue architecture to:
- Verify predicted cellular neighborhoods from computational analyses
- Confirm receptor-ligand pairs in putative interacting cells
- Validate zonated expression patterns suggested by sequencing data
Multiplex Validation: For comprehensive cell type verification, employ panels combining:
- Putative novel markers identified by scRNA-seq
- Established lineage markers for orientation
- Proteins corresponding to highly variable genes in clusters of interest
Addressing Technical Discrepancies
Several factors can contribute to apparent discrepancies between scRNA-seq and protein detection results:
- Post-transcriptional Regulation: mRNA abundance may not directly correlate with protein levels due to regulatory mechanisms
- Sensitivity Differences: scRNA-seq may detect low-abundance transcripts not reflected in protein abundance
- Spatial Sampling Variation: Different tissue sections used for sequencing versus protein detection may exhibit regional heterogeneity
- Antibody Specificity: Inadequate antibody validation can lead to false positive or negative protein detection
- Tumor Microenvironment Effects: Stromal contamination in bulk analyses can obscure cell-type-specific correlations [102]
Research Reagent Solutions
Table 3: Essential Research Reagents for Protein Validation Studies
| Reagent Category | Specific Examples | Function in Experimental Workflow |
|---|---|---|
| Validation-Qualified Antibodies | Myosin Heavy Chain isoforms (MyHC 1, 2a, 2b) [100]; Laminin (basement membrane marker) [100]; Cytokeratins (Pan-CK, CK7, CK20) [102] | Target-specific detection with verified performance in FFPE tissues |
| Fluorophore Conjugates | Alexa Fluor 350, 488, 594, 647; Texas Red; Cy dyes | Multiplex detection with minimal spectral overlap |
| Chromogenic Substrates | DAB (brown precipitate), AEC (red precipitate), Vector VIP (purple) | Enzyme-mediated color development for IHC |
| Antigen Retrieval Reagents | Tris-EDTA buffer (pH 9.0), citrate buffer (pH 6.0) | Epitope unmasking in FFPE tissues through reversal of formaldehyde cross-links |
| Blocking Reagents | Normal serum, BSA, species-specific IgG, protein-free blocking buffers | Reduction of non-specific antibody binding |
| Mounting Media | Anti-fade formulations with DAPI, permanent organic mounting media | Signal preservation and nuclear counterstaining |
| Automation Systems | Leica BOND RX [102], Akoya PhenoFusion [98] | Standardized staining protocols with run-to-run consistency |
| Image Analysis Platforms | QuPath [102], HALO, Visiopharm | Digital quantification of protein expression and spatial distribution |
Workflow Visualization
Diagram 1: Integrated Workflow for scRNA-seq Corroboration. This diagram illustrates the parallel processing of tissue samples for transcriptomic and protein-level analysis, culminating in integrated data interpretation.
Diagram 2: Parallel IHC and IF Experimental Workflows. This diagram details the shared and divergent steps in IHC and IF protocols from FFPE tissue sections, highlighting the point of methodological divergence at the detection stage.
Immunofluorescence and Immunohistochemistry provide indispensable complementary approaches for validating single-cell RNA sequencing findings at the protein level. While IHC offers superior morphological preservation and clinical translatability, IF enables multiplexed target detection that better aligns with the multi-gene expression patterns identified through scRNA-seq. The strategic integration of these protein validation techniques with transcriptomic data creates a powerful framework for comprehensive biological discovery, bridging the gap between gene expression profiling and functional protein localization within native tissue contexts. As spatial biology continues to evolve, the synergistic application of these methodologies will remain essential for authenticating novel cell types, signaling pathways, and cellular interactions revealed through high-dimensional transcriptomic technologies.
Functional validation is a critical step in translational research, bridging the gap between descriptive genomic data and therapeutic applications. While single-cell RNA sequencing (scRNA-seq) generates extensive catalogs of potential marker genes, their biological relevance and therapeutic potential remain uncertain without direct functional testing [103]. The largely descriptive nature of scRNA-seq studies produces "long descriptive ranks of markers with predicted biological function, but without functional validation, it remains challenging to know which markers truly exert the putative function" [103]. This creates a significant "valley of death" in translating academic findings to clinical applications.
Target validation is particularly crucial in drug development, as "insufficient target validation at an early stage has been linked to costly clinical failures and low drug approval rates" [103]. This protocol outlines established methodologies for gene overexpression, silencing, and knockout, providing a framework for researchers to systematically characterize candidate genes identified through scRNA-seq studies. By implementing these functional validation techniques, researchers can prioritize the most promising therapeutic targets and advance them toward clinical development.
Methodological Approaches
Comparative Analysis of Functional Validation Strategies
The table below summarizes the key characteristics, mechanisms, and applications of the three primary functional validation approaches:
Table 1: Comparison of Functional Validation Strategies
| Strategy | Description | Key Methods | Primary Applications | Considerations |
|---|---|---|---|---|
| Gene Overexpression | Increases gene expression to enhance product levels | Strong promoters, introducing extra gene copies [104] | Boosting synthesis of target products; studying gain-of-function effects [104] | May impose high metabolic burden; can disrupt natural regulatory balance |
| Gene Silencing (Attenuation) | Reduces gene expression or lowers product activity to weaken function | RNAi, siRNA, CRISPRi, antisense RNA [104] | Studying effects of reduced gene expression; fine-tuning metabolic pathways [104] | Enables precise control without complete elimination of function |
| Gene Knockout | Completely removes or deactivates a gene | CRISPR-Cas9, homologous recombination [104] | Investigating essential gene functions; eliminating specific protein production | May cause compensatory mechanisms or non-specific effects |
Research Reagent Solutions for Functional Validation
Table 2: Essential Research Reagents for Functional Validation Experiments
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Gene Editing Systems | CRISPR-Cas9, dCas9-KRAB (CRISPRi), dCas9-activator (CRISPRa) [105] | Enables gene knockout, inhibition, or activation through targeted DNA binding and modification |
| RNA-based Silencing Tools | siRNA, RNAi, shRNA, microRNA cassettes [106] [104] | Mediates sequence-specific gene silencing through mRNA degradation or translational inhibition |
| Delivery Vehicles | Lipid nanoparticles, viral vectors (lentivirus, AAV), adeno-associated viruses [107] [106] | Facilitates efficient intracellular delivery of genetic materials |
| Cell Culture Systems | Primary cells (HUVECs), organoids, stem cell technologies [103] [105] | Provides physiologically relevant models for functional testing |
| Screening Libraries | Genome-wide gRNA libraries, targeted gene sets [105] | Enables high-throughput functional genomics screens |
| Detection & Assay Systems | Single-cell RNA sequencing, FACS, viability assays, migration assays [103] [105] | Measures phenotypic outcomes of genetic perturbations |
Experimental Protocols
Protocol for Target Prioritization from scRNA-seq Data
Before embarking on functional validation, candidate genes from scRNA-seq studies must be systematically prioritized. The following workflow outlines a rigorous approach based on the GOT-IT (Guidelines On Target Assessment for Innovative Therapeutics) framework [103]:
Procedure:
Initial Gene List Compilation: Begin with top-ranking marker genes from scRNA-seq datasets. For example, start with "the top 50 most highly ranking congruent tip TEC genes" from your analysis [103].
Target-Disease Linkage Assessment (AB1): Justify biological relevance by evaluating whether the target cell phenotype is restricted to disease conditions. For instance, in tumor endothelial cell studies, note that "tip cells are restricted to TECs (99.3% of human tip cells originate from TECs)" [103].
Target-Related Safety Evaluation (AB2): Exclude markers with genetic links to other diseases. For example, "SPARC has been linked to disorders of the central nervous system, while SEMA6B has been associated with progressive myoclonic epilepsy" [103].
Strategic Novelty Consideration (AB4): Prioritize targets with minimal previous characterization in your specific context. Apply criteria such as selecting genes with "less than 20 publications vaguely describing the gene in a shared context with angiogenesis" [103].
Technical Feasibility Assessment (AB5):
- Confirm availability of perturbation tools (siRNAs, gRNAs)
- Consider protein localization (avoid secreted proteins)
- Verify cell-type specificity through analysis of "selective expression in publicly available scRNA-seq datasets" with criteria like "log-fold change >1" compared to other cell types [103].
Protocol for Gene Silencing Using siRNA Knockdown
Gene silencing through siRNA-mediated knockdown represents a versatile approach for partial gene attenuation, particularly valuable for studying essential genes where complete knockout would be lethal.
Materials:
- Primary cells relevant to your system (e.g., HUVECs for angiogenesis studies [103])
- Validated siRNA oligonucleotides (at least three different non-overlapping sequences per gene)
- Appropriate transfection reagent
- Cell culture media and supplements
- RNA/protein extraction kits
- qPCR reagents for knockdown validation
- Functional assay reagents (migration, proliferation, etc.)
Procedure:
siRNA Preparation:
- Acquire "three different non-overlapping siRNAs per gene" to control for off-target effects [103].
- Resuspend siRNAs according to manufacturer's instructions and dilute to working concentrations.
Cell Seeding and Transfection:
- Seed cells at appropriate density for your assays (e.g., 50-70% confluency for transfection).
- Transfect with siRNA complexes according to optimized protocols for your cell type.
- Include appropriate controls (non-targeting siRNA, transfection reagent only).
Knockdown Validation:
- Harvest cells 48-72 hours post-transfection for knockdown assessment.
- Validate knockdown efficiency at both RNA (qPCR) and protein (Western blot) levels.
- Select "the two siRNAs with the strongest KD efficiency for further experiments" [103].
Functional Phenotyping:
- Perform relevant functional assays based on the expected biological role:
Data Analysis:
- Normalize functional data to knockdown efficiency.
- Compare results across multiple siRNA sequences to confirm on-target effects.
- Perform statistical analysis comparing experimental and control conditions.
Protocol for CRISPR-Cas9-Mediated Gene Knockout
Complete gene knockout through CRISPR-Cas9 enables comprehensive assessment of gene function, particularly valuable for identifying essential genes and characterizing complete loss-of-function phenotypes.
Materials:
- Cas9-expressing cell line
- gRNA library (genome-wide or targeted)
- Viral packaging system (lentiviral or retroviral)
- Polybrene or similar transduction enhancer
- Selection antibiotics (puromycin, blasticidin, etc.)
- DNA extraction kit
- PCR reagents and next-generation sequencing platform
Procedure:
gRNA Library Design and Delivery:
- Design gRNAs "in silico to target either a genome-wide array of genes or specific gene sets" [105].
- Synthesize gRNAs as "chemically modified oligonucleotides and clone into a viral vector" [105].
- Package the "viral gRNA library" and transduce "a large population of Cas9-expressing cells" at appropriate multiplicity of infection (MOI) to ensure single integrations [105].
Selection and Screening:
- Apply relevant "selective pressures" based on your research question [105]:
- Drug treatments for inhibitor resistance studies
- Nutrient deprivation for metabolic gene identification
- FACS sorting "to isolate cells expressing markers indicative of specific phenotypes" [105]
- Maintain appropriate control populations (non-targeting gRNAs).
- Apply relevant "selective pressures" based on your research question [105]:
gRNA Abundance Quantification:
Hit Validation:
Protocol for Advanced Perturbomics Approaches
Integrating CRISPR screens with single-cell RNA sequencing enables high-resolution functional genomics at the single-cell level, an approach termed "perturbomics."
Materials:
- Customized CRISPR perturbation vectors with transcriptomic barcodes
- Single-cell RNA sequencing platform (10x Genomics, BD Rhapsody, etc.)
- Cell hashing antibodies for multiplexing
- Next-generation sequencing platform
- Computational resources for single-cell data analysis
Procedure:
Perturbation Library Design:
- Design gRNAs with transcriptional barcodes that can be captured during scRNA-seq.
- Consider using "CRISPRi or CRISPRa for genes where complete knockout may be lethal or for non-coding RNAs" [105].
Multiplexed Perturbation and scRNA-seq:
- Transduce cells with a pooled perturbation library at low MOI.
- Allow adequate time for phenotypic manifestation (typically 5-14 days depending on the process).
- Prepare single-cell suspensions and proceed with scRNA-seq library preparation using appropriate technology (e.g., 10x Genomics, Seq-Well, etc.).
Single-Cell Data Analysis:
- Process sequencing data to simultaneously capture:
- Cellular transcriptomes
- gRNA identities from the perturbation library
- "Identify transcriptomic changes after gene perturbation" at single-cell resolution [105].
- Cluster cells based on transcriptomic profiles and correlate with perturbations.
- Process sequencing data to simultaneously capture:
Pathway and Network Analysis:
- Perform differential expression analysis between perturbed and control cells.
- Conduct pathway enrichment analysis to identify affected biological processes.
- Construct gene regulatory networks to elucidate mechanisms.
Applications in Drug Discovery and Development
Functional validation techniques have become indispensable in modern drug discovery, particularly with the integration of single-cell technologies. Several key applications include:
Target Identification and Validation
The combination of CRISPR screens with scRNA-seq has enabled systematic identification of therapeutic targets across diverse disease areas. This "perturbomics" approach allows researchers to "identify target genes whose modulation may hold therapeutic potential for diseases such as cancer, cardiovascular disorders and neurodegeneration" [105]. These findings directly contribute "to the development of targeted drug therapies and the design of gene and cell therapies for regenerative medicine" [105].
Mechanistic Studies of Disease Pathways
Functional validation tools enable deep mechanistic investigation of disease processes. For example, in angiogenesis research, "silencing tip EC genes impairs vessel sprouting in development and disease" [103], providing both therapeutic targets and mechanistic insights. Similarly, in cancer research, CRISPR screens have been used to identify "genes that confer resistance to BRAF inhibitors, demonstrating the power of this approach in functional genomics" [105].
Therapeutic Optimization
Gene attenuation approaches provide precise tools for optimizing therapeutic interventions. In metabolic engineering, "gene attenuation allows for precise control of enzyme activity within metabolic pathways" which is "especially crucial at pathway nodes where balanced flux is needed" [104]. This principle extends to therapeutic applications where fine-tuning gene expression rather than complete knockout may yield optimal outcomes.
Troubleshooting and Technical Considerations
Common Challenges and Solutions
Table 3: Troubleshooting Guide for Functional Validation Experiments
| Challenge | Potential Causes | Solutions |
|---|---|---|
| Poor knockdown efficiency | Ineffective siRNA/gRNA, poor delivery | Use multiple siRNAs/gRNAs; optimize delivery method; validate reagents |
| High off-target effects | Sequence similarity to non-target genes | Use carefully designed controls; employ multiple distinct reagents; utilize CRISPRi with minimal off-target effects [105] |
| Cell toxicity | Overly aggressive perturbation, delivery method | Titrate perturbation intensity; use attenuated approaches (CRISPRi instead of knockout); optimize delivery conditions |
| Variable phenotypic results | Cellular heterogeneity, context dependency | Increase biological replicates; use single-cell resolution approaches; test in multiple model systems |
| Inconsistent validation | Incomplete perturbation, compensatory mechanisms | Ensure complete knockout validation; monitor for adaptive responses; use complementary approaches |
Optimization Guidelines
Cell Type Considerations: Tailor your approach to specific cell types. For difficult-to-isolate cells like neurons, "single nuclei sequencing can be beneficial" [28]. For tissues with extensive extracellular matrix, optimized dissociation protocols are essential.
Temporal Dynamics: Account for timing in phenotypic manifestation. Some phenotypes require extended time periods to develop, while others may be transient.
Validation Rigor: Always include multiple validation steps. For siRNA experiments, use "three different non-overlapping siRNAs per gene" and select "the two siRNAs with the strongest KD efficiency for further experiments" [103].
Functional validation through gene overexpression, silencing, and knockout represents a critical pathway from descriptive genomics to therapeutic applications. By implementing the protocols outlined in this document, researchers can systematically characterize candidate genes identified through single-cell RNA sequencing studies, prioritizing the most promising targets for further development. The integration of these functional validation approaches with advancing technologies—particularly CRISPR-based screens and single-cell omics—promises to accelerate the translation of genomic discoveries into clinical applications, ultimately bridging the "valley of death" between basic research and therapeutic innovation.
Conclusion
A well-executed single-cell RNA sequencing protocol is a powerful gateway to understanding biological systems at their fundamental unit. By mastering the foundational concepts, meticulously applying methodological best practices, proactively troubleshooting technical hurdles, and rigorously validating findings with orthogonal techniques, researchers can unlock profound insights into cellular heterogeneity, disease mechanisms, and developmental processes. As the field progresses, the integration of scRNA-seq with other omics technologies and spatial mapping will continue to be pivotal in advancing drug discovery, identifying novel biomarkers, and ultimately paving the way for personalized medicine approaches in clinical applications.
References
- 1. Single‐cell RNA sequencing technologies and applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 2. A practical guide to single-cell RNA-sequencing for ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0467-4]
- 3. Single-cell RNA sequencing technologies and ... [https://www.nature.com/articles/s12276-018-0071-8]
- 4. Current best practices in single‐cell RNA‐seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 5. A Strategy to Compare Single-Cell RNA Sequencing Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11457179/]
- 6. spatially aware color palette optimization for single-cell and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9272793/]
- 7. 14 Comparing and combining scRNA-seq datasets [https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/comparing-and-combining-scrna-seq-datasets.html]
- 8. Public RNA-Seq and scRNA-Seq Databases [https://bigomics.ch/blog/ultimate-guide-to-public-rnaseq-and-sc-rna-seq-databases/]
- 9. Protocol for single-cell RNA sequencing and spatial ... [https://pubmed.ncbi.nlm.nih.gov/40067823/]
- 10. Protocol for single-cell RNA sequencing and spatial ... [https://www.sciencedirect.com/science/article/pii/S2666166725000930]
- 11. Navigating single-cell RNA-sequencing: protocols, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 12. Single cell RNA-seq: An introductory overview and tools for ... [https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started]
- 13. Comparison of transformations for single-cell RNA-seq data [https://www.nature.com/articles/s41592-023-01814-1]
- 14. Single-cell RNA Sequencing: Current Progresses and ... [https://openbiotechnologyjournal.com/VOLUME/18/ELOCATOR/e18740707311249/FULLTEXT/]
- 15. Single-Cell RNA-Seq Technologies and Related ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2019.00317/full]
- 16. Single-cell full-length total RNA sequencing uncovers ... [https://www.nature.com/articles/s41467-018-02866-0]
- 17. Benchmarking full-length transcript single cell mRNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-09014-5]
- 18. FB5P-seq: FACS-Based 5-Prime End Single-Cell RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7062913/]
- 19. FIPRESCI: droplet microfluidics based combinatorial indexing ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02893-1]
- 20. Full-length mRNA sequencing resolves novel variation in 5 [https://pmc.ncbi.nlm.nih.gov/articles/PMC11794378/]
- 21. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 22. Molecular spikes: a gold standard for single-cell RNA ... [https://www.nature.com/articles/s41592-022-01446-x]
- 23. Power Analysis of Single Cell RNA-Sequencing Experiments [https://pmc.ncbi.nlm.nih.gov/articles/PMC5376499/]
- 24. What are Unique Molecular Identifiers (UMIs) and Why do ... [https://www.lexogen.com/blog/rna-lexicon-what-are-unique-molecular-identifiers-umis-and-why-do-we-need-them/]
- 25. How to Improve Your RNA-Seq Data with UMIs and ERCC ... [https://blog.genewiz.com/how-to-improve-your-rna-seq-data-with-umis-and-ercc-rna]
- 26. Single-cell RNA-seq with spike-in cells enables accurate ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02006-2]
- 27. Single-cell RNA counting using a complex set of molecular ... [https://www.rna-seqblog.com/single-cell-rna-counting-using-a-complex-set-of-molecular-spike-ins/]
- 28. Establishing single cell RNA transcriptomics: a brief guide [https://frontiersinzoology.biomedcentral.com/articles/10.1186/s12983-025-00579-x]
- 29. A robust multi-scale clustering framework for single-cell ... [https://www.nature.com/articles/s41598-025-03603-6]
- 30. a clustering guided visualization method for scRNA-seq data [https://pmc.ncbi.nlm.nih.gov/articles/PMC9048682/]
- 31. Next generation lineage tracing and its applications ... [https://www.nature.com/articles/s41540-025-00542-w]
- 32. Single Cell Isolation and Analysis - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5078503/]
- 33. Microfluidics Facilitates the Development of Single-Cell RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9312765/]
- 34. Droplet-based single-cell sequencing: Strategies and ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975024001484]
- 35. Microfluidics-free single-cell genomics with templated ... [https://www.nature.com/articles/s41587-023-01685-z]
- 36. Droplet-based single-cell RNA sequencing: decoding cellular ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06996-0]
- 37. Concepts and new developments in droplet-based single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11568944/]
- 38. Quantitative single-cell proteomics as a tool to characterize ... [https://www.nature.com/articles/s41467-021-23667-y]
- 39. Cell lysis plus depletion single-nucleus RNA-seq (CL-D ... [https://www.protocols.io/view/cell-lysis-plus-depletion-single-nucleus-rna-seq-c-g5t3by6qp.html]
- 40. Common cell lysis procedures distort ribosome profiling ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03651-1]
- 41. Functional phenotyping of genomic variants using joint ... [https://www.nature.com/articles/s41592-025-02805-0]
- 42. Single-cell nascent RNA sequencing unveils coordinated ... [https://www.nature.com/articles/s41586-024-07517-7]
- 43. Low-input and single-cell RNA sequencing [https://www.illumina.com/techniques/sequencing/rna-sequencing/ultra-low-input-single-cell-rna-seq.html]
- 44. Tutorial: guidelines for the experimental design of single- ... [https://www.nature.com/articles/s41596-018-0073-y]
- 45. Analysis of Comparative - Single ... Cell RNA Sequencing Platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC9258609/]
- 46. Systematic comparison of single - cell and... | Nature Biotechnology [https://www.nature.com/articles/s41587-020-0465-8?error=cookies_not_supported&code=215922ce-4d42-4671-9d91-c56dd4a279bb]
- 47. Research Techniques Made Simple: Single-Cell RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6363595/]
- 48. The Comparison of Single - Cell Based... RNA Sequencing Platforms [https://www.longdom.org/open-access/the-comparison-of-singlecell-rna-sequencing-platforms-based-droplets-1100795.html]
- 49. Scalable single-cell RNA sequencing from full transcripts ... [https://www.nature.com/articles/s41587-022-01311-4]
- 50. Optimized single nucleus transcriptional profiling by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9839601/]
- 51. Direct Comparative Analyses of 10X Genomics Chromium ... [https://www.sciencedirect.com/science/article/pii/S1672022921000486]
- 52. A single-cell and single-nucleus RNA-Seq toolbox for fresh ... [https://www.nature.com/articles/s41591-020-0844-1]
- 53. Comparison of whole transcriptome sequencing of fresh, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10058139/]
- 54. Structure-preserving visualization for single-cell RNA-Seq ... [https://www.nature.com/articles/s42003-023-04662-z]
- 55. emerging methods to mitigate dissociation-induced artefacts [https://www.sciencedirect.com/science/article/abs/pii/S0962892421000969]
- 56. Psychrophilic proteases dramatically reduce single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5665481/]
- 57. A single‐cell RNA labeling strategy for measuring stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9912023/]
- 58. Establishing single cell RNA transcriptomics: a brief guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403637/]
- 59. FixNCut: single-cell genomics through reversible tissue ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03219-5]
- 60. Recent advancements in tissue dissociation techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622302/]
- 61. Systematic assessment of tissue dissociation and storage ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02048-6]
- 62. Hypersonic levitation and spinning: paving the way for ... [https://www.nature.com/articles/s44172-025-00497-0]
- 63. An Optimized Tissue Dissociation Protocol for Single-Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9096112/]
- 64. snCED-seq: high-fidelity cryogenic enzymatic dissociation ... [https://www.nature.com/articles/s41467-025-59464-0]
- 65. Introducing synthetic thermostable RNase to inhibitors - single ... cell [https://pmc.ncbi.nlm.nih.gov/articles/PMC11437267/]
- 66. Robust single nucleus RNA reveals depot-specific sequencing ... cell [https://elifesciences.org/reviewed-preprints/97981]
- 67. 7 Things to Consider When Preparing a Cell Suspension ... [https://www.parsebiosciences.com/blog/cell-prep-for-single-cell-sequenci/]
- 68. Optimized protocol for isolation of high-quality single cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8605478/]
- 69. 5 tips for successful single-cell RNA-seq [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/5-tips-to-make-your-single-cell-rna-seq-experiments-a-success?srsltid=AfmBOoqWdThm9bVlsXEnnvzHT5dvpovuIEpwbiZ2z3th1Abf0aa7eqQX]
- 70. Navigating single-cell RNA-sequencing: protocols, tools ... [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00044-5]
- 71. Clinical application of single‐cell RNA sequencing in disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576031/]
- 72. Exploring and mitigating shortcomings in single-cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03525-6]
- 73. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 74. Batch correction methods used in single-cell RNA ... [https://pubmed.ncbi.nlm.nih.gov/40623818/]
- 75. Batch correction methods used in single-cell RNA sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315870/]
- 76. Integrating single-cell RNA-seq datasets with substantial ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12126-3]
- 77. Single-cell RNA sequencing reveals different cellular ... [https://www.nature.com/articles/s41523-025-00808-w]
- 78. An optimized protocol for single nuclei isolation from ... [https://www.nature.com/articles/s41598-022-14099-9]
- 79. pyComBat, a Python tool for batch effects correction in high ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05578-5]
- 80. scCausalVI disentangles single-cell perturbation ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002765]
- 81. scEVE: a single-cell RNA-seq ensemble clustering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12147100/]
- 82. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 83. Single-cell sequencing to multi-omics - Biomarker Research [https://biomarkerres.biomedcentral.com/articles/10.1186/s40364-024-00643-4]
- 84. Benchmarking multi-omics integration algorithms across ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10944570/]
- 85. scMODAL: a general deep learning framework for ... [https://www.nature.com/articles/s41467-025-60333-z]
- 86. Best practices for single-cell analysis across modalities [https://pmc.ncbi.nlm.nih.gov/articles/PMC10066026/]
- 87. A fast, scalable and versatile tool for analysis of single-cell ... [https://www.nature.com/articles/s41592-023-02139-9]
- 88. Classical Single-Cell RNA Sequencing: a comprehensive ... [https://www.lexogen.com/blog/classical-single-cell-rna-sequencing-a-comprehensive-overview/]
- 89. A Guide to Single-Cell Multiomics Data Analysis [https://www.bdbiosciences.com/en-us/learn/science-thought-leadership/blogs/single-cell-multiomics-data-analysis-guide]
- 90. Best practices for differential accessibility analysis in single ... [https://www.nature.com/articles/s41467-024-53089-5]
- 91. New cell segmentation methods for better spatial ... [https://www.fredhutch.org/en/news/spotlight/2025/10/vidd-jones-natmethods.html]
- 92. Systematic benchmarking of imaging spatial ... [https://www.nature.com/articles/s41467-025-64990-y]
- 93. Multiplexed RNA imaging and in situ profiling in living cells [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06220a?page=search]
- 94. A practical guide to spatial transcriptomics: lessons from ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925003579]
- 95. TrueProbes: Quantitative Single-Molecule RNA-FISH ... [https://elifesciences.org/reviewed-preprints/108599]
- 96. Protocol optimization improves the performance of multiplexed ... [https://datadryad.org/dataset/doi:10.5061/dryad.sj3tx96g3]
- 97. A unified pipeline for FISH spatial transcriptomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10504669/]
- 98. IHC vs IF: Key Differences, Use Cases & Which to Choose [https://www.ihisto.io/post/ihc-vs-if-comparison]
- 99. IHC vs IF: A Thorough Comparison for Research [https://www.histologix.com/insights-news/ihc-vs-if-a-thorough-comparison-for-research/]
- 100. Adaptable Immunofluorescence Protocol for Muscle Fiber ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582162/]
- 101. Integrating single-cell and spatial transcriptomics to elucidate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9888017/]
- 102. RNA sequencing and immunohistochemistry jointly ... [https://www.nature.com/articles/s41598-025-12780-3]
- 103. Prioritization and functional validation of target genes from ... [https://www.nature.com/articles/s42003-023-05006-7]
- 104. The Strategy and Application of Gene Attenuation in Metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029929/]
- 105. Perturbomics: CRISPR–Cas screening-based functional ... [https://www.nature.com/articles/s12276-025-01487-0]
- 106. CMN Weekly (8 August 2025) - Your ... [https://crisprmedicinenews.com/news/cmn-weekly-8-august-2025-your-weekly-crispr-medicine-news/]
- 107. 2025 Trends in Biotech and Life Sciences Research | Blog [https://go.zageno.com/blog/2025-trends-in-biotech-and-life-sciences-research]