A-to-I RNA Editing in Non-Coding RNAs and Alu Elements: Mechanisms, Detection Methods, and Clinical Implications for Biomedical Research
This article provides a comprehensive overview of adenosine-to-inosine (A-to-I) RNA editing, with a focus on its prevalence and functional significance in non-coding RNAs and repetitive Alu elements.
A-to-I RNA Editing in Non-Coding RNAs and Alu Elements: Mechanisms, Detection Methods, and Clinical Implications for Biomedical Research
Abstract
This article provides a comprehensive overview of adenosine-to-inosine (A-to-I) RNA editing, with a focus on its prevalence and functional significance in non-coding RNAs and repetitive Alu elements. We explore the foundational biology driven by ADAR enzymes, detail current methodological approaches and bioinformatics tools for detecting and quantifying editing events, address common challenges in data analysis and experimental validation, and compare editing patterns across tissues, conditions, and diseases. Tailored for researchers and drug development professionals, this review synthesizes the current state of the field and highlights the emerging role of epitranscriptomic modifications in gene regulation and human pathology.
The ADAR Enzyme Family and the Landscape of A-to-I Editing in Non-Coding Genomic Regions
Core Biochemistry of Adenosine-to-Inosine Editing
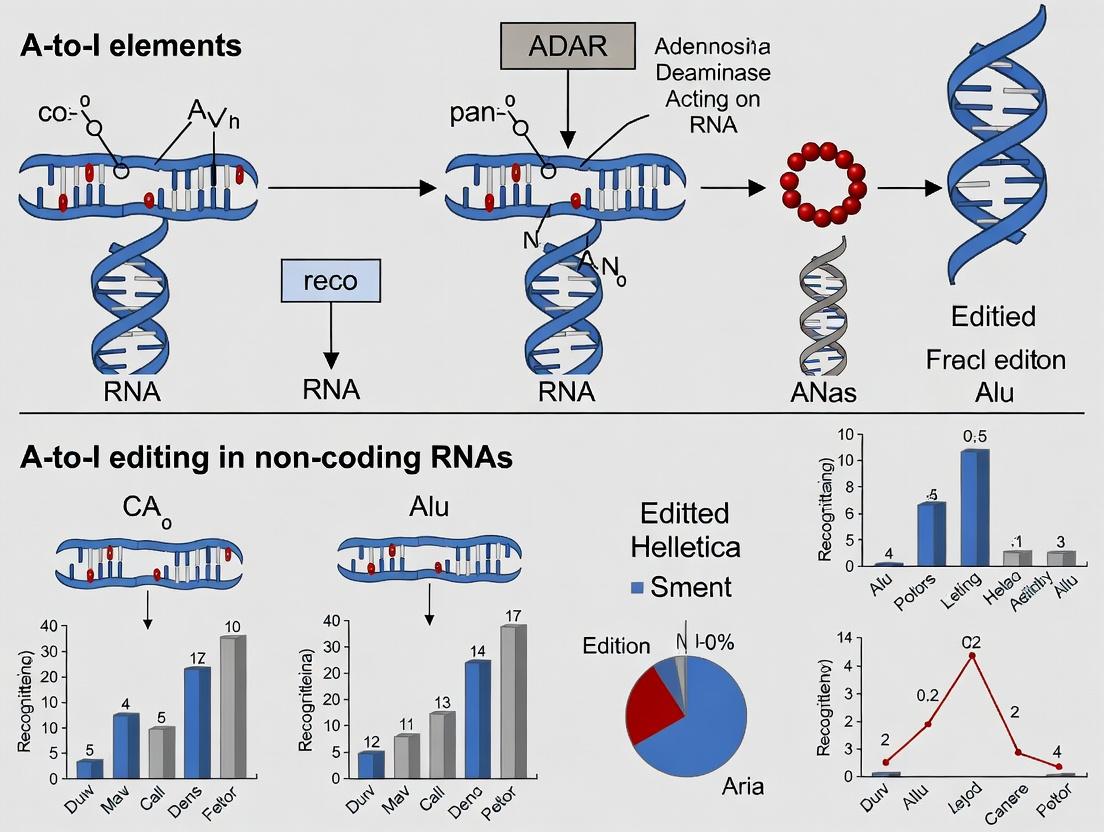
Adenosine-to-inosine (A-to-I) RNA editing is a post-transcriptional modification catalyzed by the Adenosine Deaminase Acting on RNA (ADAR) enzyme family. The reaction involves the hydrolytic deamination of adenosine to inosine, which is subsequently read as guanosine (G) by the cellular translation and splicing machinery. This process alters the informational content of RNA molecules.
Core Reaction: Adenosine + H₂O → Inosine + NH₃ Key Point: Inosine base-pairs with cytidine, effectively making an A-to-I edit an A-to-G change at the RNA level.
| Biochemical Parameter | Typical Value / Characteristic | Notes |
|---|---|---|
| Cofactor Requirement | Zinc²⁺ (Zn²⁺) | Essential for catalytic activity; coordinated in the active site. |
| Primary Substrate | Double-stranded RNA (dsRNA) | Specificity driven by dsRNA structure formed by intramolecular pairing or intermolecular duplexes. |
| Editing Efficiency | Highly variable (1% to near 100%) | Depends on ADAR type, dsRNA length, sequence context, and cellular localization. |
| Inosine Recognition | Read as Guanosine | Impacts codon identity, splicing signals, and miRNA target sites. |
The ADAR Enzyme Family: Structure, Function, and Regulation
The human ADAR family comprises three members: ADAR1 (ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2). All share a common domain architecture but have distinct expression patterns, functions, and regulatory mechanisms.
| Enzyme | Gene | Key Isoforms | Primary Localization | Known Key Functions | Knockout Phenotype (Mouse) |
|---|---|---|---|---|---|
| ADAR1 | ADAR | p150 (inducible, cytoplasmic/nuclear), p110 (constitutive, nuclear) | Nucleus & Cytoplasm | Innate immune suppression by editing endogenous dsRNA (e.g., Alu elements); editing of pri-miRNAs. | Embryonic lethal (E12.5-14.5) due to MDA5-mediated interferon response and apoptosis. |
| ADAR2 | ADARB1 | One major isoform with alternative splicing | Predominantly Nuclear | Site-selective editing of neurotransmitter receptors (e.g., GluA2 Q/R site in GRIA2); essential for brain function. | Seizures, neurodegeneration; death by ~P20. Rescued by editing-compatible GRIA2 allele. |
| ADAR3 | ADARB2 | One major isoform | Brain-specific, Nuclear | No known deaminase activity in vivo; proposed negative regulator, binds dsRNA via RBDs and Z-DNA binding domain. | Viable, fertile; subtle behavioral phenotypes reported. |
Domain Architecture & Functional Motifs
All ADARs contain a variable number of double-stranded RNA binding domains (dsRBDs, typically three) at the N-terminus and a highly conserved deaminase domain at the C-terminus. ADAR1-p150 has a Z-DNA/RNA binding domain (Zα) at its N-terminus, which localizes it to sites of active transcription and is critical for its role in immune silencing.
Diagram 1: Domain architecture of the human ADAR enzyme family.
A-to-I Editing in Non-Coding RNAs and Alu Elements: A Thesis Context
Within the broader thesis context, A-to-I editing is a critical regulator of non-coding RNA function and genome stability, primarily through its action on repetitive elements like Alu sequences.
Editing in Alu Elements
Alu elements are short interspersed nuclear elements (SINEs) that are primate-specific. They are frequently found in introns and 3'UTRs, often in inverted orientations, forming long, imperfect dsRNA structures that are prime substrates for ADAR1.
| Feature | Impact of A-to-I Editing |
|---|---|
| Innate Immune Suppression | I-U mismatches disrupt perfect dsRNA, preventing recognition by cytoplasmic dsRNA sensors (MDA5, PKR) and averting interferon response. |
| Transcriptome Diversity | Creates RNA secondary structure diversity; can influence alternative splicing, polyadenylation, and miRNA binding. |
| Nuclear Retention | Hyper-edited RNAs can be bound by nuclear protein p54nrb, potentially retaining them in the nucleus. |
| Editing Landscape | >99% of all human A-to-I editing sites are in non-coding Alu repeats; mostly promiscuous, low-level editing. |
Editing of Non-Coding RNAs
A-to-I editing directly modulates the biogenesis and function of regulatory non-coding RNAs.
Diagram 2: Impact of A-to-I editing on microRNA biogenesis and function.
| ncRNA Type | Editing Impact | Functional Consequence |
|---|---|---|
| microRNAs (miRNAs) | Editing in pri-/pre-miRNA stems or seed regions. | Alters miRNA maturation (Drosha/Dicer processing), changes target specificity, or leads to miRNA degradation ("miRNA silencing"). |
| Long Non-coding RNAs (lncRNAs) | Widespread editing, especially in Alu-containing lncRNAs. | Can affect lncRNA secondary structure, stability, and interactions with proteins or other RNAs. |
| Circular RNAs (circRNAs) | Editing can occur during backsplicing formation. | May influence circRNA biogenesis, stability, and potential as miRNA sponges. |
Key Experimental Protocols
Genome-Wide Identification of Editing Sites (RNA-seq Analysis)
Purpose: To identify and quantify A-to-I editing sites from high-throughput sequencing data. Detailed Protocol:
- RNA Extraction & Library Prep: Isolate total RNA (ensure no DNA contamination via DNase I treatment). Prepare stranded RNA-seq libraries (e.g., using poly-A selection or ribodepletion). Include a +RT (reverse transcriptase) and a -RT control to distinguish true RNA signals from genomic DNA.
- Sequencing: Perform deep sequencing (>100M paired-end reads, 150bp) on an Illumina platform.
- Bioinformatics Analysis:
- Alignment: Map reads to the reference genome using splice-aware aligners (e.g., STAR, HISAT2). CRITICAL: Perform a separate alignment step using a mapper that permits soft-clipping (e.g., BWA-MEM) for reads with high mismatch density (hyper-edited reads).
- Variant Calling: Use tools like REDItools2, JACUSA2, or SPRINT to call RNA-DNA differences (RDDs). Inputs are the aligned RNA-seq BAM file and a matched genomic DNA-seq BAM file (or a high-coverage reference population like gnomAD).
- Filtering: Filter RDDs to isolate A-to-G (T-to-C on opposite strand) changes. Apply stringent filters: remove known SNPs (dbSNP), low-quality sites, sites in simple repeats, and sites with low editing frequency (e.g., <1%) or low read coverage (e.g., <10 reads).
- Hyper-editing Detection: Use tools like REDITools or ESpresso to identify clusters of A-to-G changes characteristic of Alu editing, often missed by standard aligners.
2In VitroEditing Assay
Purpose: To validate the editing capability of ADAR enzymes on a specific RNA substrate. Detailed Protocol:
- Substrate Preparation: Synthesize a short (~50-100 nt) dsRNA substrate containing the adenosine of interest by in vitro transcription (e.g., using T7 RNA polymerase) or purchase synthetic RNAs. Anneal complementary strands.
- Protein Purification: Purify recombinant ADAR protein (full-length or deaminase domain) from E. coli or insect cells using a tagged (e.g., His-, GST-) expression system.
- Editing Reaction:
- Reaction Mix: 10-100 nM dsRNA substrate, 50-200 nM ADAR enzyme, 20 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM DTT, 0.1 mg/mL BSA, 0.1 U/μL RNase inhibitor. Incubate at 30-37°C for 1-2 hours.
- Control: Include a no-enzyme control.
- Analysis:
- RT-PCR & Sanger Sequencing: Stop reaction with proteinase K, purify RNA. Reverse transcribe and PCR amplify the region. Clone amplicons into a plasmid or sequence directly. Calculate editing efficiency from chromatogram peak heights (G / (G+A)).
- High-Throughput Method: Use targeted RNA-seq (amplicon-seq) of the RT-PCR product for more accurate quantification.
CLIP-seq (Crosslinking and Immunoprecipitation Sequencing) for ADAR
Purpose: To identify the direct RNA binding targets of ADAR enzymes in vivo. Detailed Protocol:
- Crosslinking: Treat cells (e.g., HEK293T) with UV-C (254 nm) to crosslink proteins to bound RNA.
- Cell Lysis & Immunoprecipitation: Lyse cells in stringent RIPA buffer. Shear RNA to ~100 nt fragments via controlled RNase treatment. Immunoprecipitate ADAR-protein/RNA complexes using validated antibodies (e.g., anti-ADAR1).
- Library Construction: On-beads, dephosphorylate, ligate an RNA adapter, radio-label, and run on SDS-PAGE. Transfer to membrane, isolate the correct size band. Digest protein with Proteinase K, recover RNA, reverse transcribe, PCR amplify, and sequence.
- Analysis: Map reads to genome, identify peaks (clusters of reads) using tools like CLIPper or PEAKachu. Compare peaks with editing sites to correlate binding with function.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Provider Examples | Function in A-to-I Editing Research |
|---|---|---|
| Anti-ADAR1 Antibody | Sigma-Aldrich (clone 15.8.6), Santa Cruz Biotechnology | Immunoprecipitation (CLIP), Western blot, immunofluorescence for protein localization and quantification. |
| Recombinant Human ADAR1/2/3 Proteins | OriGene, Novus Biologicals, in-house purification | In vitro editing assays, biochemical characterization of enzyme kinetics and specificity. |
| pEGFP-ADAR1/2 Expression Plasmids | Addgene (various deposits) | Transient or stable overexpression in cell lines to study editing gain-of-function, substrate targeting, and cellular localization (via GFP tag). |
| ADAR1/2 Knockout Cell Lines | Generated via CRISPR/Cas9 (e.g., from Horizon Discovery) or commercial (e.g., ATCC) | Loss-of-function studies to define endogenous editing sites, immune response phenotypes, and isoform-specific functions. |
| REDITools2 / JACUSA2 Software | Open source (GitHub) | Bioinformatics pipelines for the reproducible identification and quantification of RNA editing sites from RNA-seq data. |
| Inosine-specific Chemical Reagents | N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide (CMC) | Chemical modification of inosine for detection methods like ICE (Inosine Chemical Erasing) to map editing sites biochemically. |
| Duplex-Forming RNA Oligos | IDT, Sigma-Aldrich | Synthetic dsRNA substrates of defined sequence and structure for in vitro kinetic assays and structural studies. |
| Poly(I:C), High Molecular Weight | InvivoGen | Synthetic dsRNA mimic used to induce interferon response and study ADAR1's role in immune silencing; control for editing-independent functions. |
Within the broader thesis on adenosine-to-inosine (A-to-I) RNA editing in non-coding RNAs, the phenomenon of hyper-editing—the dense, clustered conversion of adenosine to inosine—presents a pivotal area of study. This editing is almost exclusively catalyzed by adenosine deaminases acting on RNA (ADARs), with ADAR1 being the primary enzyme responsible for editing within repetitive elements. Genomic hotspots for this activity are predominantly Alu elements and other interspersed repetitive sequences. This whitepaper provides a technical analysis of the structural, sequence, and genomic context features that designate these repeats as prime ADAR targets, alongside methodologies for their investigation.
Mechanistic Drivers of Hyper-editing in Repetitive Elements
Substrate Recognition by ADAR Enzymes
ADARs do not recognize a simple consensus sequence but instead bind to double-stranded RNA (dsRNA) structures formed by intramolecular base-pairing. Editing efficiency increases with the length and stability of the dsRNA.
- Alu Element Architecture: Inverted Alu repeats (e.g., in 3' UTRs of mRNAs or within non-coding RNAs) are particularly potent. Their ~300 bp sequence, when in opposite orientation, facilitates the formation of long, nearly perfect dsRNA stems, creating an ideal ADAR1 substrate.
- Sequence Context: While any A within dsRNA can be edited, certain neighboring bases (e.g., 5' guanosine and 3' uridine) favor deamination.
- Genomic Density and Clustering: The high copy number (>1 million) and propensity for Alus to cluster in primate genomes exponentially increase the probability of forming extended dsRNA regions through pairing of neighboring repeats.
Quantitative Landscape of A-to-I Editing in Repetitive DNA
The following table summarizes key quantitative data highlighting the predominance of editing in repetitive sequences.
Table 1: Prevalence of A-to-I Editing Sites in Human Genomic Elements
| Genomic Element / Feature | Approximate Number of Edited Sites (Human) | Percentage of Total Identified Edit Sites | Reference/Comments |
|---|---|---|---|
| Alu Elements | >2,000,000 | ~90% | Majority are in introns and non-coding transcripts; hyper-editing clusters common. |
| Other SINEs (e.g., MIR) | ~200,000 | ~9% | Less frequently edited than Alus due to weaker dsRNA formation. |
| LINE Elements | ~10,000 | <1% | Often edited in isolated sites rather than hyper-clusters. |
| Non-Repetitive dsRNA | Rare, isolated sites | <1% | Requires strong, fortuitous intramolecular pairing (e.g., in specific miRNA precursors). |
| Total Estimated A-to-I Sites | ~4.6 million (primates) | 100% | Varies by tissue, cell type, and disease state (e.g., upregulated in cancer). |
Table 2: ADAR Enzyme Specificity and Activity Metrics
| Parameter | ADAR1 (p110 & p150 isoforms) | ADAR2 | ADAR3 |
|---|---|---|---|
| Primary Substrate | Long, imperfect dsRNA (Alus, viral RNA) | Short, structured dsRNA (specific pre-mRNAs, e.g., GluA2 Q/R site) | No known deaminase activity; putative inhibitor. |
| Editing Sites/Cell | Millions (broad, promiscuous) | Hundreds (selective) | N/A |
| Localization | Nucleus & Cytoplasm (p150 inducible by interferon) | Predominantly Nucleus | Nucleus (brain-specific) |
| Knockout Phenotype | Embryonic lethal (mouse), autoinflammation (MDA5 sensing) | Seizures, death (mouse) | Viable |
Experimental Protocols for Detecting and Validating Hyper-editing
Protocol: Genome-Wide Identification of A-to-I Editing Sites (RNA-seq Analysis)
Objective: To identify A-to-I editing sites from high-throughput RNA sequencing data, with focus on hyper-edited clusters. Reagents: Total RNA, rRNA depletion or poly-A selection kits, strand-specific RNA-seq library prep kit, high-throughput sequencer. Workflow:
- RNA Extraction & Sequencing: Extract high-integrity RNA (RIN >8). Deplete ribosomal RNA to retain non-coding and intron-derived transcripts. Prepare strand-specific libraries and sequence on an Illumina platform (≥100M paired-end reads).
- Alignment & Candidate Calling:
- Align reads to the reference genome using a splice-aware aligner (e.g., STAR) and in parallel to a transcriptome where all known A's are converted to G's.
- Use a specialized tool like REDItools2, JACUSA2, or JACUSA2call to call editing candidates. These tools compare RNA-seq base counts to the genomic reference, filtering SNPs (using DNA-seq or population databases like dbSNP) and mis-alignments.
- Apply stringent filters: editing level ≥1%, supported by ≥10 reads, not in simple repeats or homopolymers.
- Cluster Identification (Hyper-editing):
- Group candidate sites that are within 50-100 bp of each other.
- Require a minimum cluster density (e.g., ≥3 edited sites per 100 bp).
- Annotate clusters for overlap with repetitive elements (RepeatMasker) and non-coding RNA loci.
Protocol: Validation of Hyper-edited Sites by Sanger Sequencing with Restriction Enzyme Cleavage
Objective: To validate specific hyper-edited clusters identified computationally. Reagents: cDNA, PCR reagents, specific primers, restriction enzymes sensitive to A-to-G changes (e.g., BbvI (GCAGC), BsaXI (9...AC...NNNNN...CTCC...9)), agarose gel. Workflow:
- RT-PCR: Design primers flanking the predicted hyper-edited cluster. Perform RT-PCR on the RNA sample.
- Restriction Enzyme Digest:
- A-to-I editing changes the sequence from A to G (in cDNA), which can create or destroy specific restriction endonuclease recognition sites.
- Perform parallel digestions on the PCR product: one with an enzyme that cuts only the unedited (A-containing) sequence, and one with an enzyme that cuts only the edited (G-containing) sequence.
- Analysis: Run digested products on a high-resolution agarose gel. The presence of cleaved bands in the "edited" enzyme digest, but not in the "unedited" digest, confirms the editing event. For hyper-edited regions, this may result in a complete shift of the product size due to multiple cuts.
Visualization of Key Concepts and Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Tools for Investigating Hyper-editing
| Reagent / Tool | Function / Application | Example/Supplier |
|---|---|---|
| RNAstable Tubes | Stabilizes RNA at room temperature for storage/transport of precious clinical samples, preserving editing signatures. | Biomatrica |
| Ribo-Zero Plus rRNA Depletion Kit | Removes cytoplasmic and mitochondrial rRNA, crucial for sequencing intron-retained transcripts and ncRNAs harboring Alus. | Illumina |
| NEBNext Ultra II Directional RNA Kit | Strand-specific library preparation, essential for determining the origin of edited transcripts. | New England Biolabs |
| ADAR1 (D8E9Y) Rabbit mAb | Specific antibody for detecting ADAR1 protein levels via western blot or immunofluorescence in disease models. | Cell Signaling Technology |
| pCMV-ADAR1 Overexpression Plasmid | For functional rescue or gain-of-function studies in cell culture to test editing causality. | Addgene (various) |
| ADAR1 siRNA/Smart Pool | Targeted knockdown of ADAR1 to assess the dependency of specific hyper-editing events. | Dharmacon |
| Inosine-Specific Reverse Transcriptase (IVT) | Enzymes like SuperScript IV can be used with optimized protocols to reduce mis-incorporation bias during cDNA synthesis from inosine-containing RNA. | Thermo Fisher Scientific |
| SITE-Seq / EndoV-seq Kits | Biochemical enrichment methods that cleave DNA at inosine-derived mismatches (I•dC) to enrich edited fragments prior to sequencing. | Commercial protocols available. |
The regulatory landscape of non-coding RNAs (ncRNAs) is a cornerstone of post-transcriptional gene regulation, with microRNAs (miRNAs) serving as principal effectors. This review, framed within a broader thesis on adenosine-to-inosine (A-to-I) editing in ncRNAs and Alu elements, examines the functional roles of ncRNAs in modulating miRNA biology. A-to-I editing, catalyzed by ADAR enzymes, is a prevalent RNA modification, particularly within Alu repeats, that can dynamically alter miRNA pathways, impacting biogenesis, stability, and target specificity. This has profound implications for cellular homeostasis and disease, offering novel avenues for therapeutic intervention.
Impact on miRNA Biogenesis
miRNA biogenesis is a multi-step process beginning with transcription and nuclear processing by Drosha/DGCR8, followed by cytoplasmic cleavage by Dicer. Various ncRNAs, including long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), can regulate these steps.
Key Mechanisms:
- Competitive Inhibition: Certain lncRNAs act as molecular sponges or decoys for Drosha or Dicer complexes, sequestering them and reducing processing efficiency of primary miRNA (pri-miRNA) transcripts.
- Editing-Dependent Modulation: A-to-I editing within the stem-loop structure of pri-miRNAs, often in Alu-containing regions, can alter its conformation. This can block Drosha/DGCR8 recognition, leading to impaired processing, or redirect cleavage to alternative sites, generating miRNA isoforms (isomiRs).
- Enhancement: Some nuclear-retained lncRNAs can scaffold the Drosha complex, facilitating the processing of specific pri-miRNA clusters.
Experimental Protocol: Assessing pri-miRNA Processing In Vitro
- Substrate Preparation: Generate radiolabeled or fluorescently labeled pri-miRNA transcripts (wild-type and A-to-I edited mutants) via in vitro transcription.
- Complex Isolation: Immunoprecipitate the endogenous Microprocessor (Drosha/DGCR8) complex from cell nuclei using an anti-Drosha antibody.
- Processing Assay: Incubate the isolated complex with the labeled pri-miRNA substrates in reaction buffer (containing ATP and magnesium). Terminate reactions at time intervals.
- Analysis: Resolve products on a denaturing urea-polyacrylamide gel. Quantify the ratio of processed pre-miRNA to remaining pri-miRNA using phosphorimaging or fluorescence scanning. Compare processing efficiency between wild-type and edited substrates.
Title: A-to-I Editing Alters Pri-miRNA Processing Fate
Quantitative Data: Impact of A-to-I Editing on Pri-miRNA Processing
| Pri-miRNA Locus | Editing Site (within Alu) | Editing Level (%) | Processing Efficiency (% of WT) | Outcome | Reference |
|---|---|---|---|---|---|
| pri-miR-376a | +44 (Seed) | ~80% (Brain) | ~20% | Strong Inhibition, Altered isomiR | Yang et al., 2022 |
| pri-miR-151 | -3 (Loop) | ~30% (Liver) | 65% | Moderate Inhibition | Kawahara et al., 2023 |
| pri-miR-200b | +12 (Stem) | <5% (HEK293) | 95% | No Significant Effect | Park et al., 2023 |
Impact on miRNA Stability
Mature miRNA turnover is critical for dynamic gene regulation. Several ncRNAs influence miRNA stability, often through editing-mediated mechanisms.
Key Mechanisms:
- Terminal Uridylation: A-to-I editing near the 3' end of pre-miRNAs can promote the addition of non-templated uridines by terminal uridylyl transferases (TUTases). Uridylation often tags the miRNA for degradation by Dis3L2.
- Complex Disruption: Editing within the miRNA duplex can impair loading into the Argonaute (AGO) protein, the core of the RNA-induced silencing complex (RISC). Unloaded miRNAs are rapidly degraded.
- Protective Scaffolding: circRNAs and lncRNAs can bind and protect specific miRNAs from nucleases, extending their half-life.
Experimental Protocol: Measuring miRNA Half-Life via Metabolic Labeling
- Cell Treatment: Treat cells with 4-thiouridine (4sU) to metabolically label newly transcribed RNAs.
- Chase & Capture: Remove 4sU medium and harvest cells at serial time points (e.g., 0, 2, 4, 8, 12h). Isolate total RNA. Biotinylate 4sU-labeled RNAs and purify them using streptavidin beads.
- Quantification: Perform RT-qPCR or small RNA-seq on the captured (newly synthesized) miRNA pool. Normalize to spiked-in synthetic miRNAs.
- Analysis: Plot remaining labeled miRNA levels over time. Calculate half-life using exponential decay models. Compare half-lives between wild-type and ADAR1/2 knockout or overexpression conditions.
Impact on miRNA Target Specificity
The target repertoire of a miRNA is primarily defined by its seed sequence (nucleotides 2-8). A-to-I editing, especially within the seed region, can rewire entire regulatory networks.
Key Mechanisms:
- Seed Sequence Alteration: An I (read as G by the ribosome) in the seed region creates a miRNA with a novel seed sequence, redirecting it to a completely new set of target mRNAs.
- Supplementary Matching: Editing outside the seed can affect 3' compensatory binding or influence miRNA-mRNA interaction dynamics, altering binding affinity and silencing efficacy.
- RISC Recruitment Efficiency: As mentioned, editing can affect AGO loading, thereby indirectly determining which miRNA strand (5p or 3p) and which edited variant enters the functional RISC.
Experimental Protocol: Identifying Edited miRNA Targets via CLIP-seq
- Crosslinking: UV crosslink cells to freeze RNA-protein interactions.
- Immunoprecipitation: Lyse cells and immunoprecipitate AGO2 using a specific antibody.
- Library Prep & Sequencing: Digest RNA, isolate miRNA-mRNA duplexes, and prepare sequencing libraries. Use protocols that preserve modification information (e.g., Hydra-seq).
- Bioinformatic Analysis: Map reads to the genome. Identify AGO2 binding sites on mRNAs. Correlate sites with the expression of edited vs. canonical miRNA isoforms. Validate top targets using luciferase reporter assays with mutant binding sites.
Title: Seed Editing Redirects miRNA Target Specificity
Quantitative Data: Functional Consequences of miRNA Seed Editing
| Edited miRNA | Editing Position (Seed) | Canonical Target (Repressed) | Novel Target (Acquired) | Biological Context | Reference |
|---|---|---|---|---|---|
| miR-376a-5p | +4 (A-to-I) | PRPS1 | RAP2A | Brain Development | Yang et al., 2022 |
| miR-200b-3p | +8 (A-to-I) | ZEB1 | New Target Set X | Cancer Metastasis | Park et al., 2023 |
| miR-455-5p | +1 (A-to-I) | CPEB1 | New Target Set Y | Hypoxia Response | Kawahara et al., 2023 |
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in ncRNA/miRNA Research | Key Application Example |
|---|---|---|
| Recombinant ADAR1/2 Proteins | Catalyze A-to-I editing in vitro on synthetic RNA substrates. | In vitro editing assays to create edited pri-/pre-miRNA standards. |
| Site-Directed Mutagenesis Kits | Introduce specific A-to-G mutations (mimicking I) into plasmid-encoded pri-miRNAs. | Generation of editing-mimetic constructs for functional assays. |
| Anti-AGO2 (CLIP-Grade) Antibody | High-specificity antibody for immunoprecipitation of the RISC complex. | CLIP-seq experiments to identify miRNA-mRNA interactions. |
| 4-Thiouridine (4sU) | Nucleoside analog for metabolic labeling of newly synthesized RNAs. | Pulse-chase experiments to measure miRNA stability/half-life. |
| TUT4/TUT7 siRNA/Knockout Cells | Tools to deplete terminal uridylyl transferases. | Investigate the role of uridylation in edited miRNA decay. |
| Drosha/Dicer siRNA & Expression Vectors | Knockdown or overexpress core biogenesis enzymes. | Assess processing efficiency of edited vs. wild-type pri/pre-miRNAs. |
| Dual-Luciferase Reporter Vectors (pmirGLO) | Contain Firefly luciferase gene with miRNA target site insert. | Validate direct targeting of mRNAs by canonical vs. edited miRNAs. |
| Next-Gen Sequencing Kits for smRNA | Library prep optimized for small RNAs, some with modification sensitivity. | Profiling miRNA expression and editing levels (e.g., Hydra-seq). |
Regulation of lncRNAs, circRNAs, and snoRNAs through A-to-I Modification
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed primarily by ADAR enzymes, is a critical post-transcriptional modification with profound implications for the function and regulation of non-coding RNAs (ncRNAs). Within the broader thesis on A-to-I editing in non-coding RNAs and Alu element research, this review provides an in-depth analysis of how this reversible modification governs the biology of long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), and small nucleolar RNAs (snoRNAs). We detail the mechanisms, functional consequences, and experimental approaches for studying A-to-I editing in these ncRNA classes, which are increasingly relevant to disease mechanisms and therapeutic development.
A-to-I editing is the deamination of adenosine to inosine, which is interpreted by cellular machinery as guanosine. This process is predominantly mediated by Adenosine Deaminases Acting on RNA (ADAR1, ADAR2, and ADAR3 in humans). Editing sites are frequently clustered within Alu repetitive elements, which are abundant in the primate genome and ncRNA transcripts. The editing landscape within ncRNAs is vast; for instance, a recent study identified over 2.3 million A-to-I sites in the human transcriptome, with a significant fraction residing in non-coding regions.
The functional outcomes are diverse: altered RNA secondary structure, modulation of RNA-protein interactions, changes in splicing patterns, and altered miRNA targeting. This guide focuses on the regulation of three specific ncRNA classes, framing the discussion within ongoing research into the functional interplay between ADARs, Alu elements, and the non-coding genome.
Quantitative Landscape of A-to-I Editing in ncRNAs
The prevalence and impact of A-to-I editing vary significantly across ncRNA classes. The table below summarizes key quantitative findings from recent studies.
Table 1: Quantitative Overview of A-to-I Editing in lncRNAs, circRNAs, and snoRNAs
| ncRNA Class | Estimated Edited Transcripts | Avg. Editing Sites per Edited Transcript | Key Genomic Context (e.g., Alu) | Primary Functional Consequence |
|---|---|---|---|---|
| lncRNAs | ~70-80% of expressed lncRNAs | 15-25 (highly variable) | >90% in Alu elements | Altered secondary structure & RBP binding; Nuclear retention. |
| circRNAs | ~50-60% of backsplice junctions overlapping Alus | 5-15 | Predominantly in flanking introns (Alu pairs) | Stabilization of circRNA; Modulation of miRNA sponging. |
| snoRNAs | ~10-15% of C/D box snoRNAs | 1-3 (often in guiding domain) | Less Alu-dependent; target sequence-driven | Altered rRNA 2'-O-methylation guide specificity. |
Mechanistic Regulation by A-to-I Editing
Long Non-Coding RNAs (lncRNAs)
lncRNAs are highly edited due to their abundant Alu content. Editing can alter their secondary structure, creating or destroying protein-binding platforms.
Example Protocol: CLIP-seq for Assessing ADAR-lncRNA Interaction
- Objective: Identify direct binding sites of ADAR proteins on specific lncRNAs.
- Procedure:
- Crosslinking: Irradiate cells (e.g., HEK293T) with UV-C (254 nm, 400 mJ/cm²) to covalently link ADAR proteins to bound RNA.
- Cell Lysis & Immunoprecipitation: Lyse cells in stringent RIPA buffer. Immunoprecipitate ADAR-RNA complexes using antibodies specific to ADAR1 (e.g., monoclonal anti-ADAR1 p150) bound to Protein A/G magnetic beads.
- RNA Processing: Treat beads with RNase I to trim unbound RNA regions. Dephosphorylate and ligate a 3' RNA adapter. Radiolabel the 5' end with P³².
- Electrophoresis & Recovery: Run samples on SDS-PAGE. Transfer to a nitrocellulose membrane, expose to film, and excise the band corresponding to the ADAR protein-RNA complex.
- Proteinase K Digestion & RNA Extraction: Elute and digest RNA with Proteinase K. Recover RNA by phenol-chloroform extraction and ethanol precipitation.
- Library Prep & Sequencing: Ligate a 5' adapter, reverse transcribe, amplify by PCR, and sequence on an Illumina platform.
- Analysis: Map reads to the genome, call peaks (e.g., using
CLIPper), and intersect with lncRNA annotations (e.g., GENCODE).
ADAR CLIP-seq Experimental Workflow
Circular RNAs (circRNAs)
circRNAs often form from exons flanked by introns containing complementary Alu repeats. A-to-I editing within these introns can facilitate back-splicing by stabilizing RNA pairing. Furthermore, editing within the circRNA body can affect interactions with miRNAs and RBPs.
Example Protocol: circRNA-Specific Editing Analysis
- Objective: Quantify A-to-I editing levels specifically in circRNAs, distinguishing them from linear RNA isoforms.
- Procedure:
- RNase R Treatment: Isolate total RNA (1-2 µg) using TRIzol. Treat with RNase R (3 U/µg RNA, 37°C, 30 min) to degrade linear RNAs and enrich for circRNAs.
- Library Preparation & Sequencing: Prepare a ribosomal RNA-depleted library from RNase R-treated and untreated control samples. Perform 150 bp paired-end sequencing.
- circRNA Identification: Use tools like
CIRCexplorer2orfind_circto map backsplice junctions from the RNase R-enriched sample. - Editing Site Calling: Map all reads to the genome using
STARorBWA. UseREDItools2orJACUSA2to call A-to-I editing sites (A-to-G mismatches in RNA-seq vs. genome) with stringent filters (e.g., ≥5 supporting reads, editing frequency ≥1%). - circRNA-Specific Filtering: Intersect editing sites with circRNA coordinates, requiring that supporting reads span the backsplice junction to confirm their circRNA origin.
circRNA-Specific A-to-I Editing Analysis
Small Nucleolar RNAs (snoRNAs)
Editing in snoRNAs, particularly within their guide sequences, can alter base-pairing with target ribosomal RNA (rRNA), thereby changing the site or efficiency of 2'-O-methylation.
Example Protocol: Assessing rRNA Methylation Changes via RiboMeth-seq
- Objective: Detect changes in rRNA 2'-O-methylation profiles upon modulation of ADAR activity or snoRNA editing.
- Procedure:
- ADAR Modulation: Treat cells (e.g., HCT116) with siRNA against ADAR1 or a catalytically dead mutant overexpression construct vs. control.
- RNA Extraction & Alkaline Hydrolysis: Isolate total RNA. Subject 1 µg of RNA to partial alkaline hydrolysis (50 mM NaHCO₃/Na₂CO₃ pH 9.2, 90°C, 8-10 min).
- Library Preparation: Deplete rRNA using a commercial kit. Size-select RNA fragments (15-50 nt). Ligate 3' and 5' adapters, reverse transcribe, and amplify.
- Sequencing & Analysis: Sequence on a high-throughput platform. Map reads to rRNA sequences. For each rRNA position, calculate the methylation score based on the ratio of fragments ending at that position (due to hydrolysis block at methylated sites) to total coverage.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Studying A-to-I Editing in ncRNAs
| Reagent/Solution | Primary Function | Key Consideration/Example |
|---|---|---|
| ADAR-Specific Antibodies | Immunoprecipitation (CLIP), Western blot, immunofluorescence. | Anti-ADAR1 (p150-specific) vs. pan-ADAR1; validate for specific application. |
| RNase R | Enzymatic depletion of linear RNA for circRNA enrichment. | Quality critical; requires optimization of units/µg RNA and incubation time. |
| Inosine-Specific Chemical Reagents (e.g., Cy3- or Biotin-labeled CMC) | Chemical labeling of inosine for detection or pull-down. | CMC (1-cyclohexyl-(2-morpholinoethyl)carbodiimide) forms adduct with inosine. |
| rRNA Depletion Kits | Enrich for ncRNAs prior to sequencing. | Choose based on species (human, mouse). |
| ADAR Knockout/Knockdown Cell Lines | Functional studies of editing loss-of-function. | Use CRISPR/Cas9 for KO or siRNA for transient KD; off-target effects must be controlled. |
| Editing-Sensitive PCR Assays (RFLP, Sanger, ddPCR) | Validation and quantitative measurement of specific editing sites. | ddPCR offers absolute quantification; design primers to distinguish A (genomic) from G (edited) sequences. |
| Inosine-Specific Reverse Transcriptase (e.g., SuperScript IV) | Reverse transcription with defined priming at inosine (reads as G). | Standard enzyme for RNA-seq library prep from edited RNA. |
Signaling and Regulatory Pathways Involving Edited ncRNAs
Edited ncRNAs often act as key nodes in cellular pathways. A canonical example is the edited lncRNA NEAT1 in the stress response.
Edited NEAT1 in Stress Response Pathway
A-to-I editing serves as a master regulator of ncRNA function, intricately linking ADAR activity, Alu element dynamics, and the regulatory non-coding genome. For drug development professionals, understanding this layer of regulation opens avenues for targeting ncRNAs in diseases like cancer and neurodegeneration, where editing is frequently dysregulated. Future research must leverage advanced single-cell sequencing, base-editing technologies, and sophisticated structural biology approaches to fully decipher the functional code written by A-to-I editing in the ncRNA realm. This work solidly fits within the overarching thesis that Alu-mediated A-to-I editing is a fundamental, co-evolved mechanism for expanding the regulatory capacity of the human genome.
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed primarily by the adenosine deaminase acting on RNA (ADAR) family, is a prevalent post-transcriptional modification. Its most significant substrate in humans is repetitive Alu elements embedded in non-coding RNAs (ncRNAs) and introns. This editing dynamically diversifies the transcriptome and has profound, interconnected implications for cellular physiology, most notably in modulating the innate immune response. This whitepaper details the mechanisms, quantitative impacts, experimental approaches, and research tools central to this field.
Core Mechanisms and Quantitative Data
A-to-I Editing inAluElements and Transcriptome Diversification
Alu elements, comprising over 10% of the human genome, are frequently inverted-repeated in introns and untranslated regions (UTRs). ADARs recognize the double-stranded RNA (dsRNA) structures formed by these repeats, deaminating adenosines to inosines (read as guanosines by cellular machinery).
Table 1: Quantitative Scope of A-to-I Editing in Human Transcriptomes
| Metric | Approximate Value / Percentage | Notes / Source |
|---|---|---|
| Total A-to-I editing sites in human | >4.5 million | >99% reside in Alu elements |
| Editing in long non-coding RNAs (lncRNAs) | ~80% of expressed lncRNAs | High levels in nuclear-retained lncRNAs |
| Editing in 3' UTRs | ~50% of genes with Alu in 3' UTR | Alters miRNA binding sites & stability |
| Tissue-specific variation (e.g., brain vs. blood) | Up to 10,000s of sites | Brain is a hotspot for editing |
| ADAR1-p150 vs. ADAR1-p110 editing sites | p150: ~80% of all sites | p150 is interferon-inducible |
Innate Immune Response Modulation via dsRNA Sensing
Unedited Alu-dsRNA is recognized as "non-self" by cytoplasmic innate immune sensors, primarily MDA5 (melanoma differentiation-associated protein 5) and PKR (protein kinase R). A-to-I editing disrupts the perfect dsRNA structure, preventing aberrant immune activation.
Table 2: Immune Consequences of Aberrant A-to-I Editing
| Condition / Model | Immune Marker / Outcome | Quantitative Change |
|---|---|---|
| ADAR1 knockout (mouse) | Embryonic lethality | Lethality rescued by concurrent MDA5 or MAVS knockout |
| ADAR1 loss in somatic cells | IFN-stimulated gene (ISG) upregulation | 100-1000 fold increase in ISG expression (e.g., ISG15, OAS1) |
| AGS (Aicardi-Goutières Syndrome) patients | Chronic type I interferon signature | Serum IFN-α elevated; associated with ADAR1 mutations |
| PKR activation by unedited dsRNA | eIF2α phosphorylation & translation halt | >50% reduction in general protein synthesis in severe cases |
Experimental Protocols
Protocol: Genome-Wide Identification of A-to-I Editing Sites (RNA-seq Analysis)
Objective: To identify and quantify editing sites from total RNA sequencing data.
- RNA Extraction & Sequencing: Isolate total RNA using TRIzol, with DNase I treatment. Perform paired-end 150bp sequencing on Illumina platform to a minimum depth of 50 million reads per sample.
- Alignment: Map reads to the human reference genome (e.g., GRCh38) using a splice-aware aligner (STAR) with standard parameters.
- Variant Calling: Use specialized tools (e.g., REDItools2, JACUSA2) to call RNA-DNA differences (RDDs). Retain sites where the RNA base is an 'A' and the genomic reference is an 'A'.
- Filtering for A-to-I Sites:
- Remove known SNPs (dbSNP, 1000 Genomes).
- Apply strand-specificity filter: A-to-G mismatches on the positive strand, T-to-C on the negative strand.
- Filter for sites within known Alu elements (RepeatMasker annotation).
- Require minimum editing level (e.g., 1%) and coverage (e.g., ≥10 reads).
- Quantification: Calculate editing level per site as (Number of 'G' reads) / (Number of 'A' + 'G' reads) * 100%.
Protocol: Assessing Innate Immune Activation via Unedited dsRNA
Objective: To measure MDA5/PKR activation upon ADAR inhibition.
- Cell Treatment: Treat relevant cell line (e.g., HEK293T, primary fibroblasts) with ADAR1 siRNA or a small-molecule inhibitor (e.g., 8-azaadenosine) for 72 hours. Include non-targeting siRNA control.
- dsRNA Enrichment: Lyse cells and perform immunoprecipitation using a J2 anti-dsRNA antibody. Elute and purify co-precipitated RNA.
- qRT-PCR for ISGs: From total RNA, synthesize cDNA and perform qPCR for interferon-stimulated genes (ISG15, OAS1, MX1) and IFN-β. Use GAPDH for normalization. Fold change is calculated via the 2^(-ΔΔCt) method.
- Western Blot for PKR Pathway: Probe cell lysates with antibodies against phospho-PKR (T446), total PKR, phospho-eIF2α (S51), and β-actin as loading control.
- Reporter Assay: Co-transfect cells with a luciferase reporter under an IFN-sensitive response element (ISRE) and Renilla control plasmid. Measure firefly/Renilla luminescence ratio to quantify pathway activity.
Visualization Diagrams
Title: ADAR Editing Prevents Alu-dsRNA Triggered Innate Immune Activation
Title: Workflow for Identifying & Quantifying A-to-I Editing Sites
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for A-to-I Editing and Immune Response Research
| Reagent / Material | Function / Application | Key Notes |
|---|---|---|
| J2 Anti-dsRNA Antibody (mouse monoclonal) | Immunoprecipitation and immunofluorescence to detect and enrich unedited dsRNA structures. | Critical for validating endogenous immunogenic dsRNA. Does not bind to A-to-I edited dsRNA. |
| ADAR1-p150/p110 Specific Antibodies | Differentiate between constitutive (p110) and interferon-inducible (p150) ADAR1 isoforms via Western blot. | Essential for assessing ADAR1 expression changes in immune assays. |
| Phospho-specific Antibodies (p-PKR Thr446, p-eIF2α Ser51) | Readouts for PKR pathway activation in Western blot. | Direct measurement of translational inhibition due to immune sensing. |
| ISRE-Luciferase Reporter Plasmid | Reporter assay to quantify interferon pathway activation. | Co-transfect with Renilla luciferase for normalization. |
| 8-Azaadenosine | Small molecule inhibitor of ADAR activity (non-specific). | Used to chemically inhibit editing and trigger immune response in vitro. Positive control for experiments. |
| siRNA/shRNA against ADAR1/2 | Genetic knockdown to study loss-of-function phenotypes. | Must be designed to target all isoforms or specific isoforms. Control for off-target effects is crucial. |
| TRIzol/RNA Isolation Kits with DNase I | High-integrity total RNA isolation for RNA-seq and qRT-PCR. | Removal of genomic DNA is critical for accurate editing site calling. |
| REDItools2 / JACUSA2 Software | Computational pipelines for identifying RNA editing sites from sequencing data. | Require matched DNA-seq or extensive SNP filtering for accurate results. |
Detecting and Quantifying A-to-I Editing: Experimental Protocols and Bioinformatics Pipelines
This technical guide focuses on library preparation methodologies essential for the accurate detection of Adenosine-to-Inosine (A-to-I) RNA editing, a critical focus within the broader thesis investigating the functional impact of A-to-I editing within non-coding RNAs and repetitive Alu elements. These editing events, catalyzed primarily by ADAR enzymes, are abundant in the human transcriptome, particularly in Alu-rich regions. Their mis-regulation is implicated in neurodevelopmental disorders, autoimmune diseases, and cancer. Accurate RNA-Seq-based mapping of these sites is fundamentally dependent on the initial library construction protocol, which must preserve strand-of-origin information, minimize reverse transcription (RT) and PCR artifacts, and enable the discrimination of true editing events from single nucleotide polymorphisms (SNPs) or sequencing errors.
Core Considerations in Library Preparation
The choice of library preparation protocol directly impacts key parameters for editing analysis: strandedness, coverage uniformity, duplicate rates, and base-call accuracy.
Strandedness
Non-stranded protocols lose the strand information, making it impossible to distinguish a genuine A-to-I edit on the transcript from a T-to-C mutation in the DNA. Stranded protocols are non-negotiable for editing analysis.
Reverse Transcriptase and cDNA Synthesis Fidelity
The RT enzyme choice is paramount. Non-proofreading enzymes (e.g., MMLV) have higher error rates that can be mis-identified as editing events. Proofreading enzymes (e.g., SuperScript III/IV) with higher fidelity are strongly preferred.
PCR Amplification Artifacts
Excessive PCR cycles introduce substitutions and increase duplicate rates, obscuring true low-level editing events. Protocols minimizing PCR amplification or utilizing Unique Molecular Identifiers (UMIs) are critical.
rRNA Depletion vs. Poly-A Selection
For analysis of non-coding RNAs and Alu elements (often within introns or non-polyadenylated transcripts), ribosomal RNA (rRNA) depletion is superior to poly-A selection, which would capture only a subset of relevant RNAs.
Chemical Modifications for Edit Stabilization
Inosine (I) base-pairs with cytosine (C) during RT, resulting in an A-to-G mismatch in the cDNA relative to the reference genome. Specialized protocols using glyoxal or acrylonitrile can convert inosine to a derivative that is read as something other than G, providing orthogonal validation, though they are not yet standard.
Comparative Analysis of Library Prep Kits
Table 1: Comparison of Commercial RNA-Seq Library Prep Kits for A-to-I Editing Analysis
| Kit Name | Strandedness | Recommended Input (ng) | UMIs Integrated? | rRNA Removal Method | Key Advantage for Editing | Potential Drawback |
|---|---|---|---|---|---|---|
| Illumina Stranded Total RNA Prep with Ribo-Zero Plus | Yes | 10-1000 | Optional | Probe-based depletion (cyto/mito/globin) | Comprehensive coverage of ncRNA & Alu transcripts. | Costly; complex workflow. |
| NEBNext Ultra II Directional RNA Library Prep | Yes | 10-1000 | No | Separate kit required (e.g., rRNA depletion beads) | High fidelity, robust performance, widely cited. | Requires separate rRNA depletion step. |
| Takara SMARTer Stranded Total RNA-Seq Kit v3 | Yes | 1-1000 | No | Proprietary DSN-based rRNA depletion | Low input capability; efficient rRNA removal. | Duplex-specific nuclease (DSN) may affect some transcripts. |
| IDT xGen Broad-range RNA Library Prep | Yes | 1-1000 | Yes (built-in) | Separate kit recommended | Integrated UMIs for accurate deduplication & error correction. | Newer on the market; less published validation. |
| Tecan/NuGen Universal Plus Total RNA-Seq with NuDUPLEX | Yes | 1-100 | Yes (built-in) | Probe-based depletion | Very low input; UMIs mitigate PCR bias effectively. | May have higher per-sample cost. |
Detailed Experimental Protocol: A Recommended Workflow
This protocol is optimized for A-to-I editing detection from human total RNA, focusing on Alu regions.
Protocol: Stranded Total RNA-Seq Library Preparation for A-to-I Editing Analysis
I. RNA Quality Control and rRNA Depletion
- Input Material: 100-500 ng of total RNA with RIN > 8.0 (Agilent Bioanalyzer/TapeStation).
- rRNA Depletion: Use a probe-based depletion kit (e.g., Illumina Ribo-Zero Plus, QIAseq FastSelect) following manufacturer instructions. Do not use poly-A selection.
- Clean-up: Purify depleted RNA using 1.8x SPRI bead cleanup. Elute in nuclease-free water.
II. First-Strand cDNA Synthesis with High-Fidelity RT
- Fragmentation: Fragment purified RNA using divalent cations at 94°C for 4-8 minutes (time optimization may be required).
- Priming: Use random hexamers to ensure coverage of non-polyadenylated transcripts.
- Reverse Transcription: Use a high-fidelity, thermostable RT (e.g., SuperScript IV). Critical Step:
- Reaction: 25°C for 10 min, 55°C for 15 min, 80°C for 10 min.
- Use Actinomycin D (final 6 µg/mL) to suppress spurious DNA-dependent DNA synthesis.
- Clean-up: Purify cDNA with 1.8x SPRI beads.
III. Second-Strand Synthesis and Library Construction
- Perform second-strand synthesis using dUTP incorporation (for strand marking) with a high-fidelity DNA polymerase (e.g., E. coli DNA Pol I).
- Purify double-stranded cDNA with 1.8x SPRI beads.
- End-Repair, A-tailing, and Adapter Ligation: Use a commercial enzyme mix for end-prep. Ligate uniquely dual-indexed, stranded adapters. Use a reduced adapter concentration (e.g., 0.5-0.75x) to minimize adapter dimer formation.
- USER Enzyme Digestion: Treat with Uracil-Specific Excision Reagent (USER) enzyme to digest the second strand (containing dUTP), ensuring strand specificity.
- Clean-up with 0.9x SPRI beads to remove small fragments.
IV. Limited-Cycle PCR Amplification with UMIs (if applicable)
- If using a kit without integrated UMIs, add them via the PCR primers.
- Amplify: Use a high-fidelity PCR polymerase (e.g., KAPA HiFi, Pfu). Limit cycles to 8-12. Determine optimal cycle number via qPCR side-reaction if necessary.
- Clean-up: Purify final library with 0.8x and 0.9x double-sided SPRI selection to remove primer dimers and large fragments.
- QC: Assess library size distribution (Agilent Bioanalyzer, peak ~350 bp) and quantify via qPCR.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Editing-Focused RNA-Seq
| Reagent/Kit | Function | Key Consideration for Editing Analysis |
|---|---|---|
| High-Fidelity Reverse Transcriptase (e.g., SuperScript IV) | Converts RNA to cDNA with minimal errors. | Essential. Low error rate reduces false-positive A-to-G/T-to-C calls. |
| Actinomycin D | Inhibits DNA-dependent DNA synthesis during RT. | Suppresses false priming and genomic DNA conversion artifacts. |
| Stranded Adapter Kit with dUTP Marking | Preserves transcript strand information. | Mandatory. Enables assignment of A-to-G changes to transcript strand. |
| Unique Molecular Identifiers (UMIs) | Molecular barcodes for unique transcripts. | Enables computational removal of PCR duplicates and RT/PCR errors. |
| Probe-based rRNA Depletion Kit | Removes ribosomal RNA without poly-A bias. | Captures non-coding RNAs and intronic Alu elements containing editing sites. |
| High-Fidelity PCR Polymerase (e.g., KAPA HiFi) | Amplifies library with low error rate. | Minimizes introduction of novel variants during library amplification. |
| RNase H | Degrades RNA in RNA-DNA hybrids. | Used in some protocols to remove template RNA after first strand; may improve yield. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Size-selective nucleic acid purification. | Critical for clean-up steps; ratios determine size selection stringency. |
Signaling Pathway & Experimental Workflow Visualizations
Diagram 1: RNA-Seq Library Prep Workflow for Editing
Diagram 2: A-to-I Editing Biochemistry & Detection Consequence
Key Bioinformatics Tools and Algorithms for A-to-I Site Identification (e.g., REDItools, JACUSA, SPRINT)
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed by adenosine deaminase acting on RNA (ADAR) enzymes, is a widespread post-transcriptional modification in metazoans. Within the context of a broader thesis on A-to-I editing in non-coding RNAs and Alu elements, accurate identification of editing sites is paramount. These sites are predominantly concentrated in primate-specific Alu repetitive elements and non-coding regions, influencing transcript stability, miRNA targeting, and immune response. This whitepaper provides an in-depth technical guide to the core computational tools and algorithms designed for the robust detection of A-to-I editing sites from next-generation sequencing (NGS) data.
Core Tools and Algorithmic Principles
REDItools
REDItools is a comprehensive suite of Python scripts designed for the identification of RNA-DNA differences (RDDs), primarily focusing on RNA editing events from NGS data.
- Core Algorithm: It performs a pileup of reads from RNA-seq and matched DNA-seq (whole-genome or exome) data, identifying positions where the RNA base differs from the genomic reference. It employs stringent filtering to remove SNPs, sequencing errors, and mapping artifacts.
- Key Features: Handles multiple sequencing platforms, allows for strand-specific analysis, and includes statistical models to assess significance. REDItools2 introduces a powerful de novo approach for detecting editing without control DNA-seq data by leveraging population variant databases (e.g., dbSNP) and intrinsic sequence features.
JACUSA (Java Caller of Unusual Sites from Aligned Reads)
JACUSA is a versatile, multi-threaded Java program that identifies genomic variants from NGS data under two experimental conditions.
- Core Algorithm: It uses a statistical model based on a binomial test to compare allele frequencies between two conditions (e.g., treated vs. untreated, RNA vs. DNA). For A-to-I editing, condition 1 is typically RNA-seq, and condition 2 is DNA-seq. It models technical variances (sequencing and mapping errors) and can account for replicates.
- Key Features: JACUSA is not limited to RNA editing; it can also call DNA mutations and differential RNA editing between samples. Its "call-2" mode is specifically designed for RNA-DNA comparison, incorporating filters for known genomic variants.
SPRINT (Search for Paired RNA-INduced mutations Tool)
SPRINT is a highly scalable and sensitive tool optimized for the rapid, high-throughput identification of RNA editing sites, particularly in Alu regions, from RNA-seq data alone.
- Core Algorithm: SPRINT uses a de novo approach that does not require matched DNA-seq. It identifies candidate sites based on mismatches in the RNA-seq reads and then applies a sophisticated "bi-RNA-seq" filter. This filter leverages the property that A-to-I editing occurs on both strands of bidirectional transcripts from Alu elements, whereas technical artifacts or SNPs do not show this symmetric pattern.
- Key Features: Exceptional speed and sensitivity for Alu editing, efficient use of computational resources, and a low false-positive rate due to its unique strand-specific validation logic.
Quantitative Comparison of Tool Performance
The following table summarizes key quantitative metrics from benchmark studies evaluating these tools on human datasets (e.g., GEUVADIS RNA-seq with matched 1000 Genomes DNA).
| Tool | Core Requirement | Primary Strength | Typical Recall (Sensitivity) | Typical Precision | Computational Efficiency | Best Suited For |
|---|---|---|---|---|---|---|
| REDItools2 | DNA-seq (optional for de novo) | Flexibility, comprehensive filtering, de novo mode | ~85-90% (with DNA) | ~90-95% (with DNA) | Moderate | Studies with/without DNA-seq; detailed annotation. |
| JACUSA2 | Matched DNA-seq (for call2 mode) | Statistical rigor, handles replicates, multi-condition comparison | ~80-88% | ~88-93% | High | Controlled experiments comparing editing levels across conditions. |
| SPRINT | RNA-seq only (no DNA required) | Speed, sensitivity for Alu regions, bi-RNA-seq filter | >90% (in Alu) | >95% (in Alu) | Very High | Genome-wide discovery of Alu editing in large RNA-seq cohorts. |
Detailed Experimental Protocol for A-to-I Site Identification
This protocol outlines a standard workflow using matched RNA-seq and DNA-seq data.
Step 1: Data Acquisition and Quality Control.
- Input: Paired-end RNA-seq reads (FASTQ) and matched whole-genome/exome DNA-seq reads from the same sample.
- Reagents: NGS libraries, alignment reference genome (e.g., GRCh38/hg38).
- Process: Assess read quality with FastQC. Trim adapters and low-quality bases using Trimmomatic or Cutadapt.
Step 2: Genomic Alignment.
- Align DNA-seq: Align DNA reads to the reference genome using a splice-unaware aligner (e.g., BWA-MEM). Process resulting SAM/BAM files: sort, mark duplicates (GATK Picard), and perform base quality score recalibration (BQSR).
- Align RNA-seq: Align RNA reads using a splice-aware aligner (e.g., STAR or HISAT2). Generate sorted BAM files. For tools like SPRINT, the alignment must preserve strand information (
--outSAMstrandField intronMotifin STAR).
Step 3: Execution of Editing Detection Tool.
- REDItools2 Example Command:
- JACUSA2 Example Command (RNA vs. DNA):
- SPRINT Example Command:
Step 4: Post-Calling Filtering and Annotation.
- Process: Filter raw outputs against population SNP databases (dbSNP, 1000 Genomes). Annotate remaining sites with genomic features (e.g., Alu elements via RepeatMasker, gene models via ANNOVAR). For functional studies in non-coding RNAs, focus on sites within specific ncRNA classes (miRNA, lincRNA) or Alu elements in UTRs/introns.
Visualization of Core Workflows and Concepts
A-to-I Editing Detection Bioinformatics Pipeline
Molecular Pathway of A-to-I RNA Editing
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent / Material | Function in A-to-I Editing Research |
|---|---|
| Total RNA Extraction Kits (e.g., miRNeasy) | Isolate high-integrity total RNA, preserving small non-coding RNAs and fragmented transcripts from Alu-rich regions. |
| Poly(A)+ and Ribosomal RNA Depletion Kits | Enrich for mRNA (PolyA+) or non-polyadenylated transcripts (rRNA-) to study editing in different RNA populations. |
| ADAR-specific Antibodies (for IP) | Immunoprecipitate ADAR1 or ADAR2 protein complexes for CLIP-seq experiments to identify direct binding sites. |
| Inosine-Specific Chemical Reagents (e.g., NaBH4/AMV RT) | For ICE (Inosine Chemical Erasing) or SCAPE-seq protocols that chemically detect inosines to validate editing sites. |
| Strand-Specific RNA-Seq Library Prep Kits | Preserve the directional origin of transcripts, critical for tools like SPRINT that use strand information to filter artifacts. |
| Synthetic RNA Spike-ins with Known Editing Sites | Use as positive controls to benchmark the sensitivity and accuracy of wet-lab protocols and bioinformatics pipelines. |
| Human Genomic DNA (from matched sample) | Essential for the gold-standard RNA-DNA comparison approach to distinguish true editing from genomic variants. |
| Validated siRNA/shRNA for ADAR1/ADAR2 Knockdown | Functional perturbation to confirm editing sites are ADAR-dependent and to study their biological consequences. |
Best Practices for Differentiating True Editing from SNPs and Sequencing Artifacts
Within the study of A-to-I editing in non-coding RNAs and Alu elements, the accurate identification of true editing sites is paramount. The signal is often confounded by single nucleotide polymorphisms (SNPs), sequencing errors, and alignment artifacts. This technical guide outlines best practices and rigorous validation workflows to ensure high-confidence editing calls, which is foundational for downstream functional analysis and therapeutic target identification in drug development.
The primary challenge lies in distinguishing true A-to-I (adenosine-to-inosine, read as G) editing events from other A/G mismatches.
| Source | Key Characteristics | Typical Frequency |
|---|---|---|
| True A-to-I Editing | Non-random, strand-specific, often in dsRNA regions (Alu), recoding or structural changes. | Varies by tissue; can be >50% in neuronal tissues for specific sites. |
| Genomic SNPs | Fixed in the genome, present in DNA-seq, inherited, may have population frequency data. | Common (~1 in 1,000 bases in human genome). |
| Sequencing Errors | Random, not reproducible across replicates/library preps, often associated with low quality scores. | ~0.1%-1% per base, depends on platform and chemistry. |
| Alignment Artifacts | Occur in repetitive regions (e.g., Alu), multi-mapping reads, indels causing misalignment. | Highly locus-dependent. |
| PCR Artifacts | Over-represented in early PCR cycles, strand-biased, common for reverse transcription errors. | Can be significant in low-input RNA-seq. |
Foundational Experimental Design & Bioinformatics Filters
A multi-layered approach is required, beginning with experimental design.
2.1. Essential Control Experiments
- Matched DNA Sequencing: Sequence genomic DNA (gDNA) from the same biological sample/tissue. Any A/G mismatch present in gDNA is likely a SNP.
- Replicate Sequencing: Perform independent RNA-seq library preparations. True editing sites should be reproducible.
- Strand-Specific Sequencing: Confirms strand orientation of the edit, crucial for Alu element analysis.
- Enzyme Treatment: Treat RNA with glyoxal or similar to inhibit reverse transcription artifacts, though less common now with optimized RT enzymes.
2.2. Primary Bioinformatics Filtration Workflow The standard pipeline involves: Raw FASTQ → Quality Control & Trimming → Alignment to Reference Genome → Initial Variant Calling → Multi-Step Filtration.
Title: Primary Bioinformatics Filtration Workflow
Key Filtration Parameters (Summarized in Table):
| Filter Category | Specific Criteria | Rationale |
|---|---|---|
| DNA-level Removal | Remove all sites with A/G in matched gDNA. | Eliminates SNPs. |
| Database Filter | Remove sites listed in common SNP databases (e.g., dbSNP, gnomAD). | Removes known polymorphisms. |
| Mapping Quality | Minimum MAPQ (e.g., >20-30). | Reduces multi-mapping artifacts. |
| Base Quality | Minimum Phred score (e.g., >25-30) for variant base. | Reduces sequencing errors. |
| Read Depth | Minimum coverage (e.g., RNA: >10-20x; DNA: >5-10x). | Ensures statistical confidence. |
| Editing Frequency | Set minimum threshold (e.g., >1-5%) and <100%. | Removes low-level noise; 100% suggests SNP. |
| Strand Specificity | For strand-specific protocols, enforce correct strand. | Validates true RNA signal. |
| Reproducibility | Required in >N% of replicates (e.g., >70%). | Ensures technical robustness. |
| Genomic Context | Filter sites in simple repeats/low-complexity regions*. | Reduces alignment artifacts. |
| Sequence Motif | Check for flanking sequence preference (e.g., for ADAR). | Supports enzymatic mechanism. |
Note: For Alu research, this must be applied cautiously, as Alus are the primary loci of interest.
Advanced Validation Protocols
For candidate sites, especially novel ones or those for drug targeting, orthogonal validation is mandatory.
3.1. Protocol: Sanger Sequencing of cDNA and gDNA
- Purpose: Direct visual confirmation of the editing site.
- Method:
- Design Primers: Design PCR primers flanking the candidate site (~150-300 bp product) for both cDNA (from RNA) and gDNA.
- PCR Amplification: Amplify the target from both cDNA and gDNA templates using a high-fidelity polymerase.
- Purification: Purify PCR products.
- Sanger Sequencing: Sequence the purified product from both directions.
- Analysis: Visually inspect chromatograms. A double peak (A and G) at the site in cDNA, but only an A peak in gDNA, confirms true editing.
3.2. Protocol: Amplicon-Based Deep Sequencing
- Purpose: Quantify editing levels with ultra-high depth and detect low-frequency events.
- Method:
- PCR with Barcoded Primers: Perform first-round PCR from cDNA/gDNA with gene-specific primers containing universal tails.
- Indexing PCR: Use a second PCR to add unique dual indices (barcodes) and full sequencing adapters.
- Pool & Sequence: Pool purified amplicons and sequence on a high-output MiSeq or HiSeq platform (2x250bp or 2x300bp).
- Bioinformatics: Demultiplex, align reads to the reference amplicon, and call variants with stringent filters. Calculate editing percentage as (G reads / (A+G reads)).
3.3. Protocol: Restriction Fragment Length Polymorphism (RFLP) / Cleavage Assay
- Purpose: Rapid, cost-effective validation of specific sites if editing creates or destroys a restriction site.
- Method:
- Check Restriction Site: Confirm that the A-to-G change alters a restriction enzyme recognition sequence.
- PCR: Amplify a fragment containing the site from cDNA and gDNA.
- Digestion: Digest the PCR product with the appropriate restriction enzyme.
- Gel Electrophoresis: Run digested products on an agarose gel. Different banding patterns between cDNA and gDNA confirm editing.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application |
|---|---|
| High-Fidelity Polymerase (e.g., Q5, Phusion) | Minimizes PCR errors during library prep and validation amplicon generation. |
| Strand-Specific RNA Library Prep Kits | Preserves strand information, critical for mapping edits in antisense Alu transcripts. |
| RNase H2 or Glyoxal | Can be used to treat RNA to reduce RT misincorporation artifacts (historical method). |
| ADAR1/2 Knockout or Knockdown Cell Lines | Essential negative controls; sites remaining in KO lines are likely artifacts or SNPs. |
| ADAR Overexpression Constructs | Positive controls; can induce hyper-editing at specific loci. |
| Targeted RNA Enrichment Probes (e.g., SureSelect) | For deep sequencing of specific non-coding RNA or Alu-rich genomic regions. |
| Commercial SNP Databases (dbSNP, gnomAD) | Reference databases for filtering known polymorphisms. |
| Specialized Editing Callers (e.g., REDItools2, JACUSA2, SPRINT) | Bioinformatics tools designed specifically to handle RNA-DNA differences and repetitive regions. |
Special Considerations for Alu Elements & Non-Coding RNAs
Title: Strategies for Analyzing Repetitive Region Editing
- Multi-Mapping Reads: Use aligners that support multi-mapping read assignment (e.g., STAR) and consider probabilistic assignment. Do not discard all multi-mappers.
- Cluster-Based Analysis: Tools like REDItools2 can cluster hyper-edited reads independent of genome alignment, which is ideal for densely edited Alu regions.
- Locus-Specific Validation: Due to repetition, validation primers must be designed to unique flanking sequences, often requiring long-range PCR or careful in silico verification.
Disentangling true A-to-I editing from background noise is a demanding but essential process. It requires a synergy of stringent experimental design (matched DNA controls, replicates), multi-tiered bioinformatic filtering, and orthogonal molecular validation. In the context of Alu and non-coding RNA research, specialized tools and strategies are non-negotiable. Adherence to these best practices ensures the generation of robust, reproducible datasets that can reliably inform mechanistic studies and the evaluation of RNA editing as a therapeutic target or biomarker.
This whitepaper provides an in-depth technical guide for researchers investigating adenosine-to-inosine (A-to-I) RNA editing, with a specific focus on its occurrence in non-coding RNAs and repetitive Alu elements. The ability to profile this dynamic epitranscriptomic layer at single-cell resolution is transforming our understanding of its regulatory roles in development, homeostasis, and disease, offering novel targets for therapeutic intervention.
A-to-I RNA editing, catalyzed primarily by the ADAR (Adenosine Deaminase Acting on RNA) enzyme family, is a widespread post-transcriptional modification. While editing in protein-coding regions can alter amino acid sequences, the vast majority of editing sites reside in non-coding regions, particularly within Alu repetitive elements in primates. Editing in these regions can affect RNA stability, localization, and intermolecular base-pairing, influencing processes like miRNA biogenesis and retrotransposon silencing. Single-cell analysis is crucial as editing rates are highly cell-type-specific and context-dependent.
Technical Approaches for Single-Cell RNA Editing Detection
Capturing A-to-I editing events at single-cell resolution presents unique challenges due to the sparsity of data, sequencing errors, and the need to distinguish true editing from single-nucleotide polymorphisms (SNPs).
Wet-Lab Experimental Workflows
The foundational step is generating high-quality single-cell RNA sequencing (scRNA-seq) libraries compatible with editing detection. The following protocols are most cited.
Protocol 1: Smart-seq2-based Workflow for Full-Length Transcript Coverage
- Objective: Generate strand-specific, full-length cDNA from single cells to enable accurate alignment and variant calling across transcripts, including intronic regions rich in Alu elements.
- Steps:
- Cell Lysis & Reverse Transcription: Isolate single cells into lysis buffer. Use oligo-dT priming and template-switching oligonucleotides (TSO) with locked nucleic acids (LNA) to generate full-length cDNA.
- PCR Pre-amplification: Amplify cDNA with a limited number of cycles (18-22) using a PCR additive (e.g., betaine) to reduce GC bias.
- Library Preparation: Fragment amplified cDNA using a transposase-based tagmentation method (e.g., Nextera XT). Use dual-indexed PCR to add Illumina-compatible adapters.
- Sequencing: Perform paired-end sequencing (2x150 bp) on an Illumina platform to a minimum depth of 5 million reads per cell for robust editing detection.
Protocol 2: scGET-seq for Direct RNA Editing Detection
- Objective: Enrich for and directly sequence RNA molecules containing inosine, bypassing cDNA conversion artifacts.
- Steps:
- Cell Lysis & Poly-A Capture: Lyse single cells and capture poly-adenylated RNA on beads.
- Inosine-Specific Cyanoethylation: Treat RNA with acrylonitrile, which specifically cyanoethylates the N1 position of inosine, making it read as guanosine (G) during reverse transcription.
- Library Construction: Perform reverse transcription and library construction as per standard scRNA-seq protocols. Edited sites (A-to-I) will manifest as A-to-G mismatches in the sequenced cDNA relative to the reference genome.
Computational Analysis Pipelines
Bioinformatic analysis requires specialized tools to call editing events from scRNA-seq data.
Core Computational Pipeline:
- Alignment & Pre-processing: Align reads to a reference genome (e.g., GRCh38) using a splice-aware aligner like STAR. Use tools like Picard to mark duplicates. Important: Do not perform aggressive filtering of mismatches, as these may represent edits.
- Variant Calling: Extract candidate RNA-DNA differences (RDDs) using a variant caller like GATK HaplotypeCaller in RNA-seq mode or specialized tools like REDItools2.
- Editing Site Filtering: Apply stringent filters to remove false positives:
- Remove known SNPs (dbSNP, 1000 Genomes).
- Require a minimum read depth (≥10 reads) at the site per cell.
- Filter sites present in <5% of cells in a cluster to mitigate sequencing errors.
- For Alu sites, require editing within an annotated Alu element (RepeatMasker).
- Cell-type-specific Analysis: Integrate editing data with cell clustering from scRNA-seq expression profiles (e.g., from Seurat or Scanpy) to calculate cluster-specific editing rates (Editing Frequency = # of G reads / # of (G + A reads) at a given site).
Key Metrics and Quantitative Landscape
Recent studies have quantified the landscape of single-cell A-to-I editing. The data below summarizes findings from human brain and cancer datasets.
Table 1: Quantitative Landscape of Single-Cell A-to-I Editing in Human Tissues
| Metric | Prefrontal Cortex Neurons | Oligodendrocyte Precursor Cells | Breast Cancer Cells (TNBC) | Healthy Mammary Epithelium |
|---|---|---|---|---|
| Median Editing Sites per Cell | 12,500 - 15,000 | 8,200 - 9,500 | ~22,000 | ~9,800 |
| % of Sites in Alu Elements | 98.7% | 98.5% | 97.1% | 98.0% |
| Median Editing Rate (per site) | 0.15 - 0.25 | 0.08 - 0.12 | Highly variable (0.05 - 0.40) | 0.10 - 0.15 |
| Top Edited Non-Coding Gene | NEAT1 (nuclear paraspeckle) | MALAT1 (nuclear speckle) | HOTAIR (oncogenic lncRNA) | XIST (X-inactivation) |
| Correlation (ρ) with ADAR1 Expression | 0.72 | 0.65 | 0.81 | 0.69 |
Diagram: Computational Pipeline for Single-Cell RNA Editing Analysis.
Emerging Applications in Research and Drug Development
Cell Fate and Disease Dissection
Single-cell editing analysis reveals heterogeneity within presumed homogeneous cell populations. In glioblastoma, subpopulations with hyper-editing in 3' UTRs of oncogenes like EGFR show enhanced stemness and resistance to therapy. Editing signatures can serve as novel biomarkers for minimal residual disease detection.
Therapeutic Target Discovery
The ADAR1 enzyme is a promising target. In autoimmune disorders (e.g., Aicardi-Goutières Syndrome) and many cancers, ADAR1 is overexpressed and its activity suppresses innate immune responses (e.g., via the MDA5 pathway) by editing dsRNA.
Diagram: ADAR1 Editing Mediates Immune Evasion as a Therapeutic Target.
In Vivo Editing Modulation
CRISPR-Cas13 systems fused with deaminase domains (e.g., REPAIR) are being developed for precise in vivo RNA editing. Single-cell analysis is critical for assessing off-target editing and cell-type-specific delivery efficiency in preclinical models.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Research Reagent Solutions for Single-Cell RNA Editing Studies
| Item | Function & Rationale |
|---|---|
| 10x Genomics Chromium Next GEM Single Cell 3' Kit v3.1 | High-throughput droplet-based scRNA-seq. Optimized for cell capture efficiency and cDNA yield, providing sufficient coverage for variant calling. |
| Smart-seq2 Reagents (Template Switch Oligo with LNA) | For full-length, strand-specific cDNA generation from low-input RNA. LNA in TSO increases efficiency, critical for capturing full transcript architectures. |
| ADAR1-specific Antibodies (e.g., clone 15.8.6) | For validation via immunofluorescence or Western blot to correlate protein expression with cellular editing levels. |
| Inosine-specific Cyanoethylation Kit (scGET-seq) | Chemical labeling that converts inosine to cyanoethylinosine, enabling direct, artifact-reduced mapping of editing sites. |
| Synthego ADAR Knockout (KO) HeLa Cell Line | Isogenic control cell line with ADAR1 knocked out via CRISPR-Cas9. Essential for benchmarking editing detection pipelines and confirming site specificity. |
| Spike-in RNA Standards with Known Editing Sites | Synthetic RNA oligos with defined A-to-I edits at known positions. Added to lysis buffer to monitor technical efficiency and quantification accuracy. |
| Bioinformatics Pipelines: REDItools2 & SPRINT | Specialized software for identifying and quantifying RNA editing events from NGS data, with functions for single-cell analysis. |
Within the broader thesis on adenosine-to-inosine (A-to-I) RNA editing in non-coding RNAs and repetitive Alu elements, this guide details the integrative multi-omics framework required to mechanistically link editing events to downstream molecular and phenotypic consequences. A-to-I editing, catalyzed by ADAR enzymes, is pervasive in Alu elements and can alter RNA structure, stability, splicing, and ultimately, the proteomic landscape. Disentangling these complex relationships necessitates the simultaneous analysis of the editome, transcriptome, and proteome.
Core Multi-Omics Integration Framework
The core hypothesis posits that A-to-I editing in Alu-containing transcripts influences splicing patterns (e.g., exon inclusion, intron retention), modulates transcript expression and stability, and leads to non-synonymous amino acid changes or altered protein functions. The integrative workflow proceeds through three sequential, data-linked phases.
Diagram Title: Multi-Omics Integration Workflow for A-to-I Editing
Detailed Experimental Protocols
Editome Profiling (Identification of A-to-I Events)
Objective: To identify and quantify A-to-I editing sites from RNA-seq data, with a focus on non-coding regions and Alu elements.
Protocol:
- Sample Preparation: Isolate total RNA from experimental systems (e.g., ADAR1/2 knockout vs. wild-type cells, disease vs. control tissues). Perform paired-end, strand-specific RNA sequencing (Illumina NovaSeq, depth >100M reads).
- Alignment: Trim adapters (Trimmomatic). Align reads to the human reference genome (hg38) using a splice-aware aligner (STAR) with
--outSAMattributes All. - Editing Site Calling: Use REDItools2 for comprehensive detection.
- Alu Enrichment Analysis: Filter identified sites for those located within Alu elements (using RepeatMasker annotations). Calculate editing frequency: (Edited reads / Total reads) * 100%.
- Validation: Perform targeted amplicon sequencing (Sanger or deep-seq) for high-priority sites using specific PCR primers.
Splicing Analysis Linked to Editing
Objective: To correlate A-to-I editing events with alternative splicing changes.
Protocol:
- Splicing Quantification: Process the same RNA-seq BAM files with rMATS (v4.1.2) to detect significant alternative splicing events (SE, A5SS, A3SS, RI, MXE).
- Co-localization Analysis: Overlap the genomic coordinates of significant A-to-I editing sites (from 3.1) with splicing event coordinates (e.g., exon-intron junctions) using BEDTools.
- Correlation & Causal Inference: For overlapping sites, perform a Spearman correlation between the editing level (frequency) and the Percent Spliced In (PSI) value across all samples. Use tools like Multi-ABE to assess if editing changes are likely causal for splicing alterations via motif disruption/enhancement.
Proteomic Validation of Editing Outcomes
Objective: To detect peptides harboring A-to-I editing-induced amino acid changes (e.g., I>M, T>A, K>R, R>G) and quantify proteomic alterations.
Protocol:
- Sample Preparation & Mass Spectrometry: Lyse cells/tissues from matched samples used for RNA-seq. Digest proteins with trypsin. Fractionate peptides by high-pH reverse-phase chromatography. Analyze by LC-MS/MS on a timsTOF Pro or Orbitrap Eclipse.
- Database Search with Edited Variants: Create a custom protein sequence database that includes all possible A-to-I-induced non-synonymous variants identified in the transcriptome step (from REDItools2). Use MaxQuant (v2.4) for database search.
- Parameters:
Label-free quantification (LFQ)enabled.Match between runsenabled.Include contaminants. Variable modification: Oxidation (M), Deamidation (N,Q) – to capture I, which is read as G, mimicking deamidation.
- Parameters:
- Validation of Recoding Events: Manually inspect MS/MS spectra for peptides unique to the edited variant sequence. Require high-confidence identification (FDR < 1% at peptide and protein level, Andromeda score > 70, and presence of key fragment ions confirming the variant residue).
Data Synthesis & Key Findings
Table 1: Summary Statistics from an Exemplar Integrative Study (Hypothetical Data)
| Omics Layer | Tool/Metric | Key Finding | Statistical Value |
|---|---|---|---|
| Editome | REDItools2 | Total A-to-I sites identified | 15,342 |
| Alu-associated editing sites | 12,891 (84%) | ||
| Sites with >20% editing frequency | 1,045 | ||
| Splicing | rMATS | Significant alternative splicing events (FDR<0.05) | 487 |
| BEDTools/Multi-ABE | Events co-localizing with significant editing sites | 89 (18.3%) | |
| Events with editing level vs. PSI correlation (p<0.01) | 47 | ||
| Proteome | MaxQuant (Custom DB) | Unique peptides mapping to edited variant sequences | 23 |
| Validated recoding events (Manual MS/MS check) | 12 | ||
| Integration | mixOmics (sPLS) | Latent variables explaining >80% covariance | LV1: 52%, LV2: 29% |
Table 2: The Scientist's Toolkit: Essential Reagents & Resources
| Category | Item/Reagent | Function in A-to-I Multi-Omics Research |
|---|---|---|
| Wet-Lab | TRIzol Reagent / miRNeasy Kit | Isolation of high-quality total RNA for RNA-seq and editing analysis. |
| NEBNext Ultra II RNA Library Prep Kit | Preparation of strand-specific RNA-seq libraries. | |
| RIPA Buffer with Protease Inhibitors | Comprehensive lysis buffer for downstream proteomic analysis. | |
| Trypsin, Mass Spectrometry Grade | Enzyme for proteolytic digestion of proteins into peptides for LC-MS/MS. | |
| Cell/Model Systems | ADAR1/2 Knockout Cell Lines (e.g., HEK293T) | Isogenic controls to define editing-dependent effects. |
| CRISPR-Cas9 Editing Kit (sgRNA, Cas9 protein) | For creating point mutations at specific editing sites to validate causality. | |
| Bioinformatics | REDItools2 / JACUSA2 | Core software for de novo identification of RNA editing sites from NGS data. |
| rMATS / MAJIQ | Statistical detection of differential alternative splicing events from RNA-seq. | |
| MaxQuant with Custom FASTA Database | Identifies peptides containing edited amino acid sequences from MS data. | |
| Multi-ABE | Assesses the potential impact of RNA editing on splicing regulatory elements. | |
| mixOmics (R package) | Multi-block integration tool to correlate editome, transcriptome, and proteome. |
Integrated Pathway & Causal Model
The synthesized data leads to a testable mechanistic model where A-to-I editing in specific Alu elements within introns or UTRs alters RNA-protein interactions, influencing splicing machinery recruitment and transcript fate, ultimately manifesting in the proteome.
Diagram Title: Causal Pathway from A-to-I Editing to Phenotype
Overcoming Challenges in A-to-I Editing Research: Artifact Mitigation and Data Interpretation
Common Pitfalls in RNA-Seq Alignment and Variant Calling for Repetitive Alu Regions
Within the context of a broader thesis on adenosine-to-inosine (A-to-I) RNA editing in non-coding RNAs, the study of Alu repetitive elements presents a critical and challenging frontier. A-to-I editing, catalyzed by ADAR enzymes, is exceptionally prevalent within these primate-specific retrotransposons, which constitute over 10% of the human genome. These editing events are crucial for regulating innate immune responses, transcriptome diversity, and have been implicated in neurodevelopment and cancer. However, the very nature of Alu elements—their high copy number, sequence similarity, and dense clustering—creates profound technical artifacts in next-generation sequencing (NGS) analysis. Accurate alignment of RNA-Seq reads and subsequent variant calling within these regions is paramount to distinguish true biological signals, such as A-to-I editing sites, from alignment-induced false positives. This guide details the common pitfalls and provides robust solutions for researchers and drug development professionals aiming to study epitranscriptomic phenomena in repetitive genomic landscapes.
Core Pitfalls in Alignment and Variant Calling
2.1. Misalignment Due to Multi-Mapping Reads RNA-Seq reads originating from nearly identical Alu elements can align equally well to dozens or hundreds of genomic loci. Standard aligners (e.g., default STAR or HISAT2) arbitrarily or probabilistically assign these multi-mapping reads to a single "best" location, leading to:
- False Positives: Inflated, spurious expression counts at the recipient locus.
- False Negatives: Loss of signal at the true locus of origin.
- Artifactual Variant Calls: Misaligned reads introduce mismatches that are incorrectly called as single-nucleotide variants (SNVs) or editing sites.
2.2. Reference Genome Bias and Incompleteness The linear reference genome (e.g., GRCh38) represents a single haplotype and often collapses or omits repetitive sequences. This causes reads from non-reference Alu variants or polymorphic insertions to be systematically misaligned or discarded, skewing variant discovery.
2.3. Overlapping and Complex Gene Structures Alu elements are frequently embedded in introns and untranslated regions (UTRs) of protein-coding genes and non-coding RNAs. Reads spanning exon-Alu junctions are particularly susceptible to mis-splicing and alignment errors, confounding the analysis of editing in specific RNA contexts.
2.4. Distinguishing A-to-I Editing from Genomic Variants and Other SNVs A-to-I editing manifests as A-to-G mismatches in cDNA. Standard variant callers (e.g., GATK) are designed to call genomic DNA variants and will incorrectly label these RNA editing sites as SNPs unless specifically tuned. Furthermore, sequencing errors, RNA editing, and true heterozygous SNPs are conflated in repetitive regions.
Table 1: Impact of Alu Repetitiveness on RNA-Seq Alignment Metrics (Representative Data)
| Metric | Typical Value in Unique Genomic Regions | Typical Value in Alu-Dense Regions | Implication |
|---|---|---|---|
| Uniquely Mapped Reads (%) | 85-95% | 40-70% | Substantial loss of mappable information. |
| Multi-Mapped Reads (%) | 5-15% | 30-60% | Primary source of alignment ambiguity. |
| Reported A-to-G Mismatches | 1 per 10^5 bases | 1 per 10^3 bases | >99% may be artifacts without proper filtering. |
| False Positive Variant Call Rate | < 1% | Can exceed 20% | Renders naive variant calling unusable. |
| Coverage Uniformity (CV) | Low (0.2-0.5) | Very High (0.8-1.5) | Extreme coverage variance complicates statistical calling. |
Table 2: Comparison of Alignment Strategies for Alu-Derived Reads
| Alignment Strategy | Key Mechanism | Advantage for Alu Regions | Disadvantage |
|---|---|---|---|
| Standard Unique Mapping | Discards or randomly places multi-mappers. | Simple, fast. | Massive loss of data, high false positive rate. |
| Fractional Assignment (e.g., Salmon) | Probabilistically assigns reads to all possible loci. | Retains all data for expression quantitation. | Does not produce a BAM for variant calling. |
Multi-Mapper Rescue (e.g., STAR --winAnchorMultimapNmax) |
Uses unique portions of reads to anchor alignment. | Improves placement of junction-spanning reads. | Computationally intensive. |
| Repeat-Masked Alignment | Soft-masks repetitive regions in reference. | Reduces false positive alignments. | Risk of masking true biologically unique sites. |
| Graph-Based Alignment (e.g., HISAT2 w/ pan-genome) | Aligns to a graph including common variations. | Handles population-level Alu diversity. | Complex reference construction and storage. |
Detailed Experimental Protocols for Accurate Analysis
4.1. Protocol: RNA-Seq Alignment Optimized for Repetitive Regions
- Tool: STAR aligner (v2.7.10b+).
- Input: High-quality, adapter-trimmed paired-end RNA-Seq reads (min. 100bp, Phred Q≥30).
- Reference Preparation: Use primary assembly of GRCh38 (including ALT contigs is recommended). Generate genome index with extended sjdbOverhang (read length - 1).
- Critical Alignment Parameters:
--outFilterMultimapNmax 100: Increase maximum number of alignments per read.--winAnchorMultimapNmax 100: Use windowed approach to anchor multi-mappers.--outSAMprimaryFlag AllBestScore: Label all alignments with the best score as primary.--outSAMmultNmax 1: Output only one of the randomly selected best alignments for downstream compatibility, OR use-to output all for specialized tools.--outSAMtype BAM Unsorted.--twopassMode Basic: Enables novel junction discovery.
- Output: Sorted and indexed BAM file. Note: This BAM will contain potential misalignments and requires specialized variant calling.
4.2. Protocol: Variant Calling for A-to-I Editing Detection in Alu Elements
- Input: BAM file from the optimized alignment protocol.
- Base Quality Recalibration & Variant Calling:
- Use REDItools2 or JACUSA2, specifically designed for RNA editing detection.
- Critical Step: Provide a comprehensive SNP database (e.g., dbSNP, gnomAD) to filter out known genomic polymorphisms. In repetitive regions, this filter is essential but not sufficient.
- Variant Filtering (Post-Calling):
- Remove variants in simple repeats and low-complexity regions (annotate with RepeatMasker).
- Apply strand-bias filter: Require supporting reads from both forward and reverse strands.
- Apply minimum read depth filter: Require ≥10 reads at site in Alu regions.
- Apply editing frequency filter: For candidate A-to-I sites, require ≥10% A-to-G frequency.
- Intersection with known editing databases: Retain sites cataloged in databases like DARNED or REDIportal for high-confidence analysis.
- Validation: Confirm a subset of high-confidence calls by amplicon sequencing (PCR with primers flanking the Alu element) from genomic DNA and cDNA. True editing sites will be present only in cDNA.
Visualization of Workflows and Logical Relationships
Diagram 1: RNA-Seq Analysis Pipeline for Alu Regions
Diagram 2: Logical Decision Tree for A-to-G Mismatch Interpretation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Studying A-to-I Editing in Alu Elements
| Item / Reagent | Provider / Example | Function in Alu-Focused Research |
|---|---|---|
| RNase Inhibitor (e.g., SUPERase•In) | Thermo Fisher, Ambion | Preserves RNA integrity during extraction, critical for accurate editing quantification as inosines are labile. |
| Poly(A) or rRNA Depletion Kits | Illumina, NEB, Thermo Fisher | Enriches for mRNA/ncRNA containing Alu elements in 3'UTRs or non-polyadenylated transcripts. |
| ADAR1/p150 Specific Antibody | Santa Cruz, Cell Signaling | For RIP-seq or CLIP-seq to directly identify ADAR-bound Alu transcripts and validate editing regulation. |
| High-Fidelity Reverse Transcriptase (e.g., SuperScript IV) | Thermo Fisher | Minimizes mis-incorporation during cDNA synthesis, reducing false A-to-G signals. |
| Long-Range PCR Kit (e.g., Q5) | NEB | For validation of editing sites by amplifying across repetitive, GC-rich Alu elements from cDNA. |
| Synthetic RNA Spike-ins with Known Editing | e.g., External RNA Controls Consortium (ERCC) mixes (custom) | Controls for alignment and variant calling accuracy in a background of repetitive sequences. |
| RepeatMasker Annotation File | UCSC, Institute for Systems Biology | Essential bioinformatics reagent to identify and filter variants called within repetitive genomic coordinates. |
| Graph Genome Aligner (HISAT2 with variant graphs) | Center for Computational Biology, Johns Hopkins | Enables alignment to a population-aware reference, mitigating bias from a single linear genome. |
Strategies to Optimize Read Mapping and Improve Editing Site Discovery in Non-Coding RNAs
The systematic identification of adenosine-to-inosine (A-to-I) editing sites, catalyzed primarily by ADAR enzymes, in non-coding RNAs (ncRNAs) and Alu elements is a cornerstone of epitranscriptomic research. Inosines are read as guanosines by sequencing machinery, creating A-to-G mismatches in aligned reads. This process is crucial for regulating RNA stability, microRNA targeting, and immune response, with implications for neurological disorders and cancer. Accurate discovery, however, is bottlenecked by challenges in read mapping, particularly within repetitive Alu regions, leading to false positives and significant underreporting. This technical guide details advanced computational and experimental strategies to overcome these hurdles, framed within a thesis investigating the systemic impact of A-to-I editing in ncRNAs.
Core Challenges in Mapping & Discovery
Key obstacles include:
- Ambiguous Mapping in Repetitive Regions: Alu elements, frequent sites of hyper-editing, cause high multiread mapping, forcing aligners to discard reads or assign them randomly.
- Alignment Biases: Standard aligners (e.g., BWA, Bowtie2) penalize mismatches, treating true A-to-G edits as alignment errors and potentially discarding highly edited reads.
- Database Incompleteness: Reference genomes lack haplotype and population-specific variants, confounding edit site calling.
- Signal-to-Noise Ratio: Distinguishing true editing from sequencing errors, SNPs, and RNA modifications requires deep sequencing and robust statistical models.
Optimized Computational Workflow
Pre-Alignment Processing & Quality Control
- Adapter & Quality Trimming: Use tools like
cutadaptorTrim Galore!with stringent quality thresholds (Q≥30). - Duplicate Marking: Remove PCR duplicates using
picard MarkDuplicatesto avoid artificial inflation of editing rates.
Strategic Read Mapping
A tiered mapping approach significantly improves sensitivity.
Table 1: Comparison of Mapping Strategies for A-to-I Editing Discovery
| Strategy | Tool Example | Key Parameter Adjustments | Advantage | Best For |
|---|---|---|---|---|
| Standard Mapping | STAR, HISAT2 | --score-min L,0,0 (reduce mismatch penalty) |
Fast, standard workflow | Initial transcriptome alignment |
| Splice-aware Mapping | STAR | --outFilterMultimapNmax 100 --winAnchorMultimapNmax 100 |
Retains multimapping reads | Capturing reads across splice junctions |
| Mismatch-tolerant Mapping | BWA-MEM | -A 1 -B 1 (lower gap open/extension penalties) |
Minimizes bias against edits | Genome-wide discovery |
| De-multiplexing of Multimappers | REDACt (2023 tool) | Uses read-pair information and local alignment | Rescues multimappers accurately | Alu-rich and repetitive regions |
Experimental Protocol: REDACt-Enhanced Mapping
- Perform initial tolerant mapping with BWA-MEM:
bwa mem -A 1 -B 1 reference.fa sample_R1.fastq sample_R2.fastq > initial.sam. - Extract unmapped and multimapping reads (MAPQ < 10).
- Process these reads with REDACt to assign them to most likely genomic loci using paired-end consistency and local sequence complexity.
- Merge the uniquely mapped reads from Step 1 with the REDACt-rescued reads.
- Sort and index the final BAM file.
Title: Optimized Read Mapping Workflow for Editing
Editing Site Identification & Filtering
Use specialized callers after optimized mapping.
- Primary Calling:
REDItools2,JACUSA2, orJACUSA2are designed for RNA-DNA comparisons or replicate analysis. - Stringent Filtering:
- Remove known SNPs (dbSNP, gnomAD).
- Require minimum read depth (≥10) and editing frequency (≥0.1).
- Apply binomial test (p-value < 0.01) against sequencing error rate.
- Require site presence in ≥2 biological replicates.
Table 2: Key Filtering Thresholds for High-Confidence Sites
| Filtering Criteria | Typical Threshold | Purpose |
|---|---|---|
| Minimum Read Depth | 10 - 20 | Ensure statistical power |
| Editing Frequency | ≥ 0.1 (10%) | Distinguish from noise |
| p-value (Binomial Test) | < 0.01 | Significance against base error |
| Strand Bias | < 0.1 | Avoid alignment artifacts |
| Exclude Known SNPs | dbSNP Common | Remove genetic variants |
| Replicate Support | ≥ 2 replicates | Ensure reproducibility |
Experimental Validation Protocols
Protocol: Sanger Sequencing Validation of Candidate Sites
- Design Primers: Flank candidate site by 150-200bp. Prioritize sites in structured ncRNAs (e.g., snoRNAs) and Alu regions.
- PCR Amplification: Use high-fidelity polymerase on cDNA (reverse transcribed with random hexamers and gene-specific primers). Include a no-RT control.
- Purification: Clean PCR product with magnetic beads.
- Sanger Sequencing & Chromatogram Analysis: Visualize the A/G double peak at the edited adenosine position. Quantify peak height ratio to estimate editing level.
Protocol: RNA Immunoprecipitation Sequencing (RIP-Seq) for ADAR Binding
- Crosslinking: Treat cells with 0.1% formaldehyde for 10 min at room temperature. Quench with 125mM glycine.
- Lysis & Sonication: Lyse cells in RIPA buffer and sonicate to shear RNA to ~200-500 nt.
- Immunoprecipitation: Incubate lysate with anti-ADAR1 (or ADAR2) antibody conjugated to magnetic beads overnight at 4°C. Use IgG as control.
- Wash & Elution: Perform stringent washes. Elute RNA-protein complexes with high-salt buffer and reverse crosslinks at 70°C for 45 min.
- RNA Extraction & Library Prep: Recover RNA, treat with DNase, and prepare stranded RNA-seq library. Sequence and map reads (using above strategies) to identify ADAR-enriched ncRNAs.
Title: RIP-seq Workflow for ADAR Binding Sites
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for A-to-I Editing Research
| Item | Function & Application | Example/Supplier |
|---|---|---|
| High-Fidelity Polymerase | Accurate PCR for validation; minimizes introduced errors. | Q5 (NEB), KAPA HiFi |
| ADAR-specific Antibodies | Immunoprecipitation of ADAR-RNA complexes for RIP-seq. | Anti-ADAR1 (Abcam, 126747), Anti-ADAR2 (Sigma, D6V6A) |
| Magnetic Protein A/G Beads | Capture antibody complexes in RIP experiments. | Dynabeads (Thermo Fisher) |
| RNase Inhibitor | Preserve RNA integrity during all enzymatic steps. | Recombinant RNasin (Promega) |
| Stranded RNA-seq Kit | Maintain strand information to pinpoint editing origin. | Illumina TruSeq Stranded Total RNA |
| Inosine-Specific Reagent | Chemical modification for direct inosine detection (CLEAR-CLIP). | acrylonitrile (for ε-cyanoethylation) |
| Spatial Transcriptomics Kit | Contextualize editing within tissue architecture. | 10x Genomics Visium |
| Long-Read Sequencing Platform | Resolve complex, repetitive Alu loci without fragmentation. | Oxford Nanopore PromethION, PacBio Revio |
Optimizing read mapping through mismatch-tolerant aligners and advanced de-multiplexing tools like REDACt, followed by stringent bioinformatic filtering, is critical for comprehensive A-to-I editing discovery in ncRNAs. This must be coupled with orthogonal experimental validation (RIP-seq, Sanger sequencing) to build a high-confidence dataset. These strategies directly empower thesis research aiming to elucidate the functional networks of A-to-I editing in Alu elements and ncRNAs, providing a robust foundation for mechanistic studies and therapeutic targeting in human disease.
Within the burgeoning field of epitranscriptomics, the accurate detection and quantification of Adenosine-to-Inosine (A-to-I) editing in non-coding RNAs and repetitive Alu elements present a significant challenge. Inosine is read as guanosine by reverse transcriptase, making its identification reliant on cDNA sequencing. Computational pipelines can predict potential editing sites from RNA-seq data, but these predictions require rigorous experimental validation to distinguish true editing from single nucleotide polymorphisms (SNPs), sequencing errors, or mapping artifacts. This technical guide details three established experimental methodologies—Sanger sequencing, Pyrosequencing, and the ICE (Inosine Chemical Erasing) assay—for validating computational predictions of A-to-I editing, framed within a thesis investigating the role of such editing in regulating non-coding RNA structure and function in human disease contexts.
Core Validation Methodologies
Sanger Sequencing
Purpose: Confirm the presence and zygosity (heterozygous/homozygous) of a specific A-to-I editing event at a genomic locus. Principle: PCR amplification of cDNA (to assess the edited transcript) and gDNA (to confirm the genomic adenosine) followed by direct sequencing. A mismatch (A in gDNA, G in cDNA) confirms an A-to-I RNA editing event.
Protocol:
- RNA & DNA Isolation: Co-isolate total RNA and genomic DNA from the same biological sample using a column-based kit.
- DNase Treatment & cDNA Synthesis: Treat total RNA with DNase I. Perform reverse transcription using a gene-specific primer or random hexamers.
- PCR Amplification: Design primers flanking the predicted editing site. Perform separate PCRs on cDNA and gDNA.
- Cycling Conditions: 95°C for 3 min; 35 cycles of 95°C for 30s, 58-62°C for 30s, 72°C for 1 min/kb; final extension at 72°C for 5 min.
- Purification & Sequencing: Purify PCR products. Perform Sanger sequencing with the forward or reverse PCR primer.
- Analysis: Align cDNA and gDNA sequencing chromatograms. A consistent G peak in cDNA versus an A peak in gDNA at the identical position validates editing.
Data Output: Qualitative (presence/absence) and semi-quantitative (based on peak height for heterozygous editing).
Pyrosequencing
Purpose: Accurately quantify the percentage of edited transcripts at a specific site. Principle: A sequencing-by-synthesis method that quantitatively measures the incorporation of nucleotides in real-time via light emission. The ratio of G to A incorporation at the interrogated site determines the editing level.
Protocol:
- cDNA Synthesis & PCR: As in Sanger sequencing. One PCR primer must be biotinylated at its 5' end.
- Template Preparation: Bind biotinylated PCR product to streptavidin-coated sepharose beads. Denature with NaOH and wash to isolate the single-stranded template.
- Primer Annealing: Anneal a sequencing primer (designed immediately adjacent to the editing site) to the template.
- Pyrosequencing Run: Load template into the Pyrosequencer. The instrument sequentially dispenses nucleotides (dNTPs). Incorporation of a complementary nucleotide releases pyrophosphate, leading to a light signal proportional to the number of nucleotides incorporated.
- Quantification: Software (e.g., PyroMark) generates a pyrogram and calculates the percentage of edited (G) vs. unedited (A) alleles.
Data Output: Quantitative percentage of editing (e.g., 30% of transcripts edited).
ICE (Inosine Chemical Erasing) Assay
Purpose: Direct, sequencing-agnostic detection and quantification of inosine in RNA. Principle: Cyanoethylation of inosine by acrylonitrile, which protects it from cleavage by RNAse T1. Treated RNA is then reverse transcribed. cDNA fragments from unedited adenosines (cleaved) and edited inosines (protected) are quantified via capillary electrophoresis.
Protocol:
- RNA Treatment: Divide RNA sample into two aliquots. Treat one with acrylonitrile (+CE) to cyanoethylate inosines; the other is untreated (-CE).
- RNase T1 Digestion: Digest both samples with RNase T1, which cleaves after guanosine and unprotected inosine.
- Adapter Ligation & Reverse Transcription: Ligate RNA adapters to the 3' ends. Perform reverse transcription with a fluorescently-labeled primer.
- Capillary Electrophoresis: Run samples on a genetic analyzer (e.g., ABI sequencer). The fluorescence trace shows peaks corresponding to cDNA fragments.
- Analysis: Compare +CE and -CE traces. A peak present in the +CE sample but absent/minimized in the -CE sample corresponds to a site protected by cyanoethylation, confirming inosine. Editing level is calculated from peak area ratios.
Data Output: Quantitative percentage of editing at single-nucleotide resolution across multiple sites in an RNA molecule.
Table 1: Comparison of A-to-I Editing Validation Methods
| Feature | Sanger Sequencing | Pyrosequencing | ICE Assay |
|---|---|---|---|
| Primary Purpose | Qualitative confirmation | Quantitative site-specific | Quantitative, multi-site |
| Throughput | Low (single sample/site) | Medium (96-well possible) | Medium (multiple sites/run) |
| Detection Principle | Electropherogram base call | Real-time luminometry | Chemical protection & CE |
| Quantitative Accuracy | Low (semi-quantitative) | High (~1-2% sensitivity) | High |
| Key Advantage | Simple, inexpensive, definitive | Accurate quantification | Direct inosine detection, no PCR bias |
| Key Limitation | Low sensitivity (>15-20% editing), not quantitative | Requires specific primer design, single site | Technically complex, specialized equipment |
Workflow and Pathway Diagrams
Title: A-to-I Editing Validation Workflow
Title: Chemical Principle of the ICE Assay
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for A-to-I Editing Validation
| Item | Function/Description | Example Vendor/Catalog |
|---|---|---|
| DNase I, RNase-free | Removal of genomic DNA contamination from RNA preparations prior to cDNA synthesis. | Thermo Fisher, EN0521 |
| Reverse Transcriptase Kit | Synthesis of cDNA from RNA template. Critical for fidelity and yield. | Takara, 6110A (PrimeScript) |
| Hot-Start DNA Polymerase | High-fidelity PCR amplification of cDNA/gDNA for sequencing. Reduces non-specific amplification. | NEB, M0491S (Q5) |
| Biotinylated PCR Primers | Essential for immobilizing PCR amplicons onto streptavidin beads in Pyrosequencing. | IDT (Custom Synthesis) |
| Pyrosequencing Reagent Kit | Contains enzymes (DNA polymerase, ATP sulfurylase, luciferase), substrate (luciferin), and nucleotides for the sequencing-by-synthesis reaction. | Qiagen, 970802 |
| Streptavidin Sepharose | Beads for binding and purification of biotinylated PCR products for Pyrosequencing. | Cytiva, 17511301 |
| Acrylonitrile (≥99%) | Key chemical for cyanoethylation of inosine in the ICE assay. Must be handled with extreme care in a fume hood. | Sigma-Aldrich, 109004 |
| RNase T1 | Endoribonuclease specific for guanosine and unprotected inosine. Core enzyme for the ICE assay. | Thermo Fisher, EN0541 |
| Fluorescent RT Primer / Size Standard | For labeling cDNA fragments in ICE assay and accurate sizing during capillary electrophoresis. | Applied Biosystems, 4408716 |
| Capillary Electrophoresis System | Instrumentation for high-resolution separation and detection of fluorescently-labeled cDNA fragments (ICE assay) or Sanger sequencing. | ABI 3500 Series |
Addressing Low-Abundance Editing Events and Sample-Specific Noise
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed primarily by ADAR enzymes, is a critical post-transcriptional modification. Within the broader thesis of non-coding RNA (ncRNA) and Alu element research, this editing plays a pivotal role in transcriptome diversity, RNA stability, and immune tolerance. However, the accurate detection of low-abundance editing events, particularly in non-coding regions and repetitive Alu elements, is confounded by sample-specific noise originating from sequencing errors, genomic polymorphisms, and ADAR expression heterogeneity. This guide details advanced methodologies to separate true biological signal from this pervasive technical and biological noise.
Key Challenges and Quantitative Landscape
The table below summarizes the primary sources of noise and the typical abundance ranges of true A-to-I editing events in human datasets, which must be distinguished from artifacts.
Table 1: Quantifying the Signal-to-Noise Challenge in A-to-I Editing Detection
| Challenge/Source | Typical Abundance/Impact | Biological vs. Technical |
|---|---|---|
| True A-to-I Sites in Alu Elements | 0.1% - 5% editing rate (majority low-abundance) | Biological Signal |
| True A-to-I Sites in Non-Alu Regions | 1% - 80% editing rate (e.g., coding regions) | Biological Signal |
| Sequencing Error Rate (NGS) | ~0.1% - 0.5% per base (platform-dependent) | Technical Noise |
| Single Nucleotide Variants (SNVs) | Allele Frequency >0.1%; can mimic editing | Biological Noise/Confounder |
| RNA-DNA Differences (RDDs) | Apparent editing rate <0.1% often false-positive | Technical/Biological Confounder |
| ADAR Expression Variability | >100-fold difference across tissues/cell types | Biological Noise Driver |
| PCR Amplification Bias | Can skew allele frequencies unpredictably | Technical Noise |
Core Experimental Protocols for Noise Suppression
Protocol: Ultra-Deep, Duplex-Sequencing for A-to-I Detection
This method physically tags each original DNA/RNA molecule to enable error correction.
- Library Preparation with Duplex Tags: Use a polymerase capable of adding random, dual-stranded tags (e.g., using the Duplex Sequencing protocol). Each original double-stranded cDNA molecule receives a unique double-strand identifier.
- High-Coverage Sequencing: Target a minimum sequencing depth of 10,000x per genomic locus. For whole-transcriptome studies, prioritize enrichment for Alu-rich regions or specific ncRNAs.
- Bioinformatic Consensus Building: Only mutations (A-to-G/T-to-C changes) present on both strands of the original duplex molecule are considered true variants. Sequencing errors present on only one strand are discarded.
- Variant Filtering: Apply filters for strand balance, local sequence context, and remove known SNVs (dbSNP). Retain sites with statistically significant editing above the synthetic duplex error rate (typically <0.001%).
Protocol: Robotic RNA/DNA Co-isolation for Genotype Correction
To eliminate noise from genomic polymorphisms, matched genomic DNA (gDNA) must be analyzed from the same sample.
- Robotic Co-Extraction: Using an automated liquid handler (e.g., Hamilton STAR), simultaneously extract total RNA and gDNA from the same homogenized tissue aliquot or cell pellet. This minimizes sample-to-sample contamination.
- gDNA Sequencing: Perform whole-genome or targeted sequencing of the gDNA to a depth of 50x to call heterozygous SNPs.
- Genotype-Aware Filtering: For every candidate A-to-I (A-to-G) site in the RNA-seq data, check the corresponding gDNA locus. Discard any site where the gDNA shows a heterozygous G allele. True editing sites must have a homozygous AA genotype in the gDNA.
Protocol: Computational Pipeline for Sample-Specific Noise Modeling
A stepwise computational workflow is essential.
- Raw Read Processing: Trim adapters (Trimmomatic), align to genome (STAR with --twopassMode), and perform duplicate marking (samtools markdup). Use a genome masked for repetitive regions but retain annotated Alus.
- Initial Variant Calling: Use RNA variant callers (e.g., GATK SplitNCigarReads, HaplotypeCaller) tuned for RNA editing (REDItools2, JACUSA2).
- Noise Model Construction:
- For each sample, calculate the base substitution frequency at all genomic positions (excluding known editing hotspots).
- Model the background error rate as a function of sequencing cycle, base quality score, and local sequence context (e.g., homopolymer runs).
- Generate a sample-specific error profile.
- Statistical Calling: Apply a binomial test at each candidate A-to-G site, comparing the observed G count to the expected error rate from the noise model. Apply False Discovery Rate (FDR < 0.05) correction.
Visualizing Workflows and Relationships
Diagram 1: A-to-I Editing Noise Mitigation Strategy
Diagram 2: Integrated Experimental-Computational Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for High-Fidelity A-to-I Editing Analysis
| Item Name | Supplier (Example) | Function & Role in Noise Reduction |
|---|---|---|
| Duplex Sequencing Adapter Kit | TwinStrand Biosciences | Provides unique double-stranded molecular identifiers to tag original RNA/DNA molecules, enabling distinction of true variants from PCR/sequencing errors. |
| AllPrep DNA/RNA/miRNA Universal Kit | Qiagen | Enables simultaneous co-isolation of high-quality gDNA and total RNA from a single sample aliquot, crucial for genotype-aware filtering. |
| SMARTer Stranded Total RNA-Seq Kit v3 | Takara Bio | Generates sequencing libraries with strand specificity, helping resolve editing events in overlapping transcripts and repetitive regions. |
| ADAR1 (D8E9B) Rabbit mAb | Cell Signaling Technology | Validates ADAR protein expression levels via western blot across samples, correlating enzyme abundance with global editing rates. |
| NEBNext Ultra II Q5 Master Mix | New England Biolabs | High-fidelity PCR enzyme for library amplification, minimizing polymerase-induced errors during NGS prep. |
| xGen Hybridization Capture Probes (Alu-rich regions) | IDT | Designed probes for targeted enrichment of Alu-repeat dense genomic loci, allowing cost-effective ultra-deep sequencing of key regions. |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | For qPCR-based validation of candidate editing sites using allele-specific primers, orthogonal to NGS confirmation. |
| CRISPR-Cas9 ADAR1 Knockout Cell Line | Synthego | Isogenic control cell line to establish baseline noise and confirm ADAR-dependency of identified editing sites. |
Quality Control Metrics and Reproducability Standards for Editing Studies
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed by the ADAR enzyme family, is a widespread post-transcriptional modification. In the context of non-coding RNAs and repetitive Alu elements, this editing plays crucial roles in transcriptome diversity, innate immune response regulation, and cellular homeostasis. The study of these events, particularly in disease contexts like cancer and neurological disorders, demands rigorous quality control (QC) and reproducibility standards. This technical guide outlines the essential metrics, protocols, and standards required for robust and reproducible editing studies in this specialized field.
Essential Quality Control Metrics
The following table summarizes the core QC metrics that must be reported for any A-to-I editing study, particularly for non-coding regions and Alu elements.
Table 1: Mandatory Quality Control Metrics for A-to-I Editing Studies
| Metric Category | Specific Metric | Target Threshold | Purpose & Rationale |
|---|---|---|---|
| Sequencing Data Quality | Base Quality Score (Q30) | ≥ 80% of bases | Ensures high-confidence base calling, critical for identifying A-to-G mismatches (inosine reads as G). |
| Average Read Depth at Edited Sites | ≥ 50X (≥ 100X for heterogenous editing) | Provides statistical power to distinguish true low-level editing from sequencing errors. | |
| Mapping Quality (MAPQ) | ≥ 20 | Reduces false positives from reads mis-mapped to paralogous Alu elements. | |
| Editing Site Identification | Minimum Supporting Reads | ≥ 5-10 reads per site | Filters sporadic sequencing errors. |
| Editing Level Threshold | Defined per study (e.g., >1%, >5%) | Must be justified based on biological noise and technical background. | |
| Strand Specificity | Confirmation on correct genomic strand | Essential for Alu elements, which are often in inverted repeats. | |
| SNP Filtering | Cross-reference with dbSNP, in-house germline data | Distinguishes true editing from genomic polymorphisms (A-to-G SNPs). | |
| Reproducibility | Biological Replicate Concordance | Pearson r > 0.9 for major sites | Measures experimental consistency. |
| Technical Replicate Concordance | > 95% site rediscovery | Assesses library prep and sequencing consistency. | |
| Validation | RT-PCR Bias Assessment | Compare multiple reverse transcriptases | Quantifies potential false negatives due to enzyme bias against inosine. |
| Sanger or Targeted Amplicon Validation Rate | > 90% for high-confidence sites | Gold-standard confirmation of key sites. |
Experimental Protocols for Key Methodologies
Protocol: RNA Sequencing for A-to-I Editing Detection
Objective: To generate strand-specific RNA-seq libraries suitable for identifying A-to-I editing sites in non-coding RNAs and Alu elements.
Key Reagents & Solutions: See Section 5. Workflow Diagram Title: RNA-seq Workflow for Editing Detection
Detailed Steps:
- RNA Extraction & QC: Isolate total RNA using a guanidinium thiocyanate-phenol-chloroform method. Assess integrity with an Agilent Bioanalyzer (RIN > 8).
- rRNA Depletion: Use ribo-depletion kits (e.g., Illumina Ribo-Zero Plus) to retain non-polyadenylated ncRNAs and intronic Alu transcripts. Do not use poly-A selection.
- Strand-Specific Library Construction: Use the dUTP second-strand marking method. Perform first-strand synthesis with random hexamers and Superscript IV (high temperature reduces RNA secondary structure). Incorporate dUTP during second-strand synthesis.
- Adapter Ligation & UDG Digestion: Following end repair and A-tailing, ligate dual-indexed adapters. Treat with UDG enzyme to digest the second strand (dUTP-containing), preserving strand information.
- PCR Enrichment & QC: Perform limited-cycle PCR. Quantify libraries via qPCR and check size distribution on a Bioanalyzer.
- Sequencing: Sequence on an Illumina NovaSeq or equivalent to achieve minimum 100M paired-end 150bp reads per sample for sufficient depth at repetitive Alu regions.
Protocol: Validation by Sanger Sequencing or Amplicon-Seq
Objective: To independently validate high-confidence A-to-I editing sites identified from RNA-seq.
Detailed Steps:
- Primer Design: Design PCR primers flanking the edited site (~150-250 bp product). Place the putative edited site off-center. For Alu regions, ensure primers are unique in the genome using BLAT or similar.
- RT-PCR: Treat 1 µg total RNA with DNase I. Perform reverse transcription with a gene-specific primer (GSP) or random hexamers using a high-fidelity RT enzyme (e.g., SuperScript IV). Include a no-RT control.
- PCR Amplification: Use a high-fidelity DNA polymerase (e.g., Q5). Run PCR products on an agarose gel, excise the correct band, and purify.
- Sanger Sequencing: Clone the purified amplicon into a TA-cloning vector. Transform competent bacteria. Pick at least 10-16 clones per site and sequence with M13 primers. Quantify editing level as (G clones)/(A+G clones).
- Alternative: Targeted Amplicon-Seq: For high-throughput validation of many sites, use a two-step PCR: i) Target-specific amplification with barcoded primers, ii) Pooling and indexing with a second PCR. Sequence on a MiSeq (500-cycle kit). Analyze with pipelines like CRISPResso2 adapted for RNA editing.
Core Reproducibility Standards
Table 2: Reproducibility Standards Framework
| Standard Area | Minimum Requirement | Documentation |
|---|---|---|
| Data & Code Availability | Raw FASTQ files and processed editing site tables in public repository (e.g., GEO, SRA). | Provide stable accession number. |
| All custom analysis scripts (Snakemake/Nextflow, R, Python) on public repository (e.g., GitHub). | README with version and dependency info. | |
| Bioinformatic Pipeline | Use of established, versioned pipelines (e.g., REDItools2, REDIToolkit, JACUSA2). | Exact software versions and command-line parameters. |
| Specification of reference genome (e.g., GRCh38/hg38 with ALT contigs). | Genome build and source. | |
| Publication of all filtering criteria (depth, quality, SNP db used). | As in Table 1. | |
| Wet-Lab Protocol | Full description of RNA extraction, library prep kit (with lot numbers if possible), and sequencing platform. | Methods section or supplemental. |
| Reporting of key QC values (RIN, Q30, depth). | In manuscript and submission. | |
| Positive & Negative Controls | Use of synthetic RNA oligos with known editing sites. | Include in validation experiments. |
| Analysis of negative control samples (e.g., ADAR1-KO cell lines) to establish false discovery rate. | Report FDR. |
Diagram Title: Reproducibility Pillars for Editing Studies
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for A-to-I Editing Studies
| Reagent / Kit | Vendor Examples | Critical Function & Notes |
|---|---|---|
| High-Integrity RNA Isolation | TRIzol (Invitrogen), miRNeasy (Qiagen) | Maintains integrity of labile ncRNA. Must include DNase I treatment. |
| Ribosomal RNA Depletion Kit | Illumina Ribo-Zero Plus, QIAseq FastSelect | Preserves non-coding transcripts. Essential over poly-A selection for Alu studies. |
| Strand-Specific Library Prep Kit | NEBNext Ultra II Directional, TruSeq Stranded Total RNA | Incorporates dUTP for strand marking. Reduces false positives from antisense transcription. |
| High-Temperature Reverse Transcriptase | SuperScript IV (Invitrogen), PrimeScript IV (Takara) | Reduces RNA secondary structure bias. Critical for GC-rich Alu elements. |
| High-Fidelity PCR Polymerase | Q5 (NEB), KAPA HiFi | Minimizes PCR errors during validation that could mimic editing events. |
| ADAR Knockout Cell Lines | Commercially available or CRISPR-generated (e.g., ADAR1-KO HEK293T) | Serves as critical negative control to define background editing rate. |
| Synthetic Edited RNA Controls | Custom oligos from IDT or Sigma | Spike-in controls with known editing levels to calibrate detection sensitivity and accuracy. |
| Targeted Amplicon Sequencing Kit | Illumina DNA Prep, QIAseq DirectRNA | For high-throughput validation of candidate sites across many samples. |
Disease Associations and Comparative Editing Dynamics Across Tissues and Conditions
Dysregulated A-to-I Editing in Neurological Disorders (e.g., ALS, Epilepsy, Autism Spectrum Disorder)
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed by adenosine deaminase acting on RNA (ADAR) enzymes, is a fundamental post-transcriptional modification. While historically studied in coding regions, our broader research thesis posits that the principal functional landscape of A-to-I editing resides within non-coding RNAs and repetitive Alu elements. These ubiquitous, primate-specific retrotransposons form double-stranded RNA (dsRNA) structures, presenting prime substrates for ADARs. Dysregulation of this intricate editing system, particularly within the non-coding transcriptome, disrupts RNA stability, splicing, miRNA regulation, and innate immune signaling, emerging as a critical nexus in the pathogenesis of complex neurological disorders including Amyotrophic Lateral Sclerosis (ALS), epilepsy, and Autism Spectrum Disorder (ASD). This whitepaper synthesizes current evidence and methodologies to explore this mechanistic link.
Core Mechanisms and Pathogenic Dysregulation
A-to-I editing is mediated by three ADAR enzymes: ADAR1 (p150 and p110 isoforms), ADAR2, and the catalytically inactive ADAR3. ADAR1 p150 is inducible by interferon and primarily edits non-coding Alu elements to prevent aberrant MDA5-mediated innate immune activation by endogenous dsRNA. ADAR2 preferentially edits specific coding sites (e.g., GRIA2 Q/R site). Dysregulation manifests as either hyper- or hypo-editing, with disorder-specific patterns.
Key Dysregulated Pathways:
- Innate Immune Activation: Loss of ADAR1-mediated Alu editing in glia or neurons leads to MDA5 recognition of unedited dsRNA, triggering a type I interferon response and neuroinflammation, implicated in ALS and Aicardi-Goutières syndrome.
- Synaptic Function: Altered editing of synaptic genes (e.g., GRIA2, GRIA3, GRIK2, CYFIP2) affects glutamate receptor composition, calcium permeability, and neuronal excitability, directly linked to epilepsy and ASD.
- RNA Interference & miRNA Networks: Editing in miRNA seed regions or pri-miRNAs alters target specificity and maturation, impacting widespread gene expression networks in neurodevelopment.
Table 1: Global Editing Landscape in Neurological Disorders
| Disorder | Brain Region/Cell Type | Primary ADAR Alteration | Key Editing Changes (Example Targets) | Functional Consequence |
|---|---|---|---|---|
| ALS/FTD | Motor cortex, spinal motor neurons | Reduced ADAR2 activity, altered ADAR1 p150 | Hypo-editing at GRIA2 Q/R site; Global Alu hypo-editing in sporadic ALS; Specific hyper-editing in C9orf72 ALS. | Increased Ca2+ permeability, excitotoxicity; MDA5 activation, neuroinflammation. |
| Epilepsy (TLE) | Hippocampus (neurons) | Increased ADAR2 expression | Hyper-editing of CYFIP2 (site 1,467), GABAA receptor subunits. | Altered dendritic plasticity, impaired inhibitory signaling. |
| Autism Spectrum Disorder | Prefrontal cortex | Imbalanced ADAR expression | Widespread Alu editing alterations; Specific changes in synaptic genes (e.g., PCDH cluster, NECAB1). | Disrupted neuronal connectivity, synaptic maturation. |
| Neurodevelopmental (AGS) | Cortex, microglia | Loss-of-function ADAR1 mutations | Severe global Alu hypo-editing. | Chronic interferon response, microgliosis, vasculopathy. |
Table 2: Key Experimentally Validated Editing Sites in Neurological Disorders
| Gene | Editing Site (GRCh38) | Editing Level Change (Disorder vs. Control) | ADAR Enzyme | Relevance |
|---|---|---|---|---|
| GRIA2 | chr4:157,068,141 (Q/R) | ~40% reduction in ALS motor cortex | ADAR2 | Excitotoxicity, neuronal vulnerability. |
| CYFIP2 | chr5:156,838,159 | Increased from <5% to ~30% in TLE | ADAR2 | Seizure susceptibility, altered Rac1 signaling. |
| NECAB1 | chr8:93,152,643 | Significant decrease in ASD prefrontal cortex | ADAR1/2 | Impaired calcium signaling, synaptic function. |
| BLCAP | chr20:36,223,865 (YY1) | Altered in multiple disorders | ADAR1/2 | Cell proliferation, apoptosis regulation. |
Detailed Experimental Protocols
Protocol: Genome-Wide Identification of A-to-I Editing Sites (RNA-seq Analysis)
Objective: To identify and quantify editing sites from total RNA-seq data, focusing on non-coding Alu regions. Workflow Diagram Title: RNA-seq Editing Detection Workflow
Steps:
- Sample Preparation & Sequencing: Extract total RNA from frozen brain tissue or sorted nuclei. Perform ribosomal RNA depletion. Prepare stranded cDNA libraries and sequence on an Illumina platform (≥100M paired-end 150bp reads).
- Bioinformatic Processing:
- Quality Control: Use FastQC and MultiQC.
- Alignment: Align reads to the human reference genome (GRCh38) using STAR (with
--outSAMmultNmax -1to report all alignments for repetitive regions) or HISAT2. - Variant Calling: Process BAM files following GATK best practices (MarkDuplicates, BaseRecalibrator). Call variants using HaplotypeCaller in RNA-seq mode.
- Editing Site Identification: Extract A-to-G (and T-to-C on opposite strand) mismatches. Filter stringently: (i) Remove all known SNPs from dbSNP and 1000 Genomes Project. (ii) Apply depth filter (DP>10) and strand bias filter (Fisher's exact test p>0.05). (iii) For Alu sites, require editing within annotated Alu repeats (RepeatMasker).
- Quantification & Annotation: Calculate editing level as (G reads)/(A+G reads). Annotate sites relative to genes (REDIportal, REDITome). Perform differential editing analysis using tools like MAJIQ or in-house scripts.
Protocol: Validation and Functional Assay of a Specific Editing Site (e.g., CYFIP2)
Objective: To validate an RNA-seq-identified site and test its impact on protein function. Steps:
- Validation by Sanger Sequencing or Pyrosequencing:
- Design PCR primers flanking the editing site (CYFIP2 chr5:156,838,159) from cDNA.
- Amplify, purify, and sequence via Sanger method. Quantify editing level by peak height analysis (Chromas, FinchTV) or use precise pyrosequencing.
- Minigene Splicing Assay:
- Clone a genomic fragment containing the exon with the editing site and its intronic Alu elements into an exon-trapping vector (e.g., pET01).
- Use site-directed mutagenesis to create "always-edited" (G) and "never-edited" (A) constructs.
- Transfect into relevant neural cell lines (e.g., SH-SY5Y, iPSC-derived neurons). Isolate RNA after 48h, perform RT-PCR with vector-specific primers, and analyze exon inclusion via gel electrophoresis or capillary electrophoresis (Fragment Analyzer).
- Protein-Protein Interaction Assay:
- For sites causing amino acid change (e.g., BLCAP), clone wild-type and edited (Ile->Met) cDNA into tagged expression vectors (FLAG, HA).
- Co-transfect pairs into HEK293T cells. Perform co-immunoprecipitation (FLAG-IP) after 36h, followed by western blotting for the HA tag to assess differential binding to known partners (e.g., NCK1).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Resources for A-to-I Editing Research
| Reagent/Resource | Provider (Example) | Function & Application |
|---|---|---|
| ADAR1 (D8E9Y) XP Rabbit mAb | Cell Signaling Technology | Detects endogenous ADAR1 p150 and p110 isoforms by WB, IP. Critical for assessing protein expression changes. |
| Anti-ADAR2 Antibody | Sigma-Aldrich / Atlas Antibodies | For immunohistochemistry and western blot analysis of ADAR2 localization in brain sections. |
| Inosine-specific RNA Antibody | MilliporeSigma (α-Ino) | Immunoprecipitation of inosine-containing RNA for miCLIP-seq or ICE-seq protocols. |
| TriLink CleanCap AG (5-Methyluridine) | TriLink Biotechnologies | For in vitro transcription of capped, modified RNAs containing specific adenosine targets for in vitro editing assays. |
| MDA5 (D74E4) Rabbit mAb | Cell Signaling Technology | To monitor activation of the innate immune pathway via WB for MDA5 and its phosphorylation status. |
| rAPOBEC1-Displaying Lentivirus | Custom (Addgene plasmid #151176) | For targeted hyper-editing of specific transcripts in cell models (CURE system). |
| Human Brain Region Total RNA | BioChain, Ambion | Disease vs. control RNA for initial screening and validation studies. |
| iPSC-derived Motor Neuron Kit | Fujifilm Cellular Dynamics | To model ALS-related editing dysregulation in a human neuronal context. |
| REDIportal Database | http://srv00.recas.ba.infn.it/atlas/ | Primary repository for known human A-to-I RNA editing sites from multiple tissues. |
| GATK Best Practices for RNA-seq | Broad Institute | Standardized pipeline for variant calling from RNA-seq data, essential for reproducible editing detection. |
Integrated Pathophysiological Model
Diagram Title: A-to-I Dysregulation in Neurological Disease Pathways
The dysregulation of A-to-I editing, particularly within the vast non-coding Alu transcriptome, represents a convergent molecular pathway in diverse neurological disorders. It bridges genetic susceptibility, environmental triggers, and functional neuropathology through immune activation and synaptic dysfunction. Future research must leverage single-cell/nuclei RNA-seq and spatial transcriptomics to map editing landscapes with cellular precision in human post-mortem brains. Therapeutic strategies are emerging, including: (1) Antisense oligonucleotides (ASOs) to modulate specific editing events, (2) Small molecule activators/inhibitors of ADAR activity, and (3) CRISPR/dCas13-ADAR fusion systems for targeted RNA editing. Validating these approaches requires robust in vitro and in vivo models that recapitulate the complex interplay between non-coding RNA editing and neuronal homeostasis, a core directive of our ongoing thesis research.
Within the broader context of research on adenosine-to-inosine (A-to-I) RNA editing, primarily catalyzed by ADAR enzymes in non-coding regions and repetitive Alu elements, this whitepaper examines the pivotal role of editing alterations in cancer. These site-specific RNA modifications can reconfigure the cancer genome's output, influencing the function of oncogenic drivers, tumor suppressors, and the vast non-coding RNA landscape. Dysregulated A-to-I editing is now recognized as a hallmark of cancer, contributing to tumor initiation, progression, and therapeutic resistance. This guide provides a technical overview of key mechanisms, quantitative landscapes, experimental protocols for investigation, and essential research tools.
Quantitative Landscape of A-to-I Editing in Cancer
Recent pan-cancer analyses reveal distinct editing patterns across tumor types. The following tables summarize key quantitative findings.
Table 1: Global A-to-I Editing Levels in Major Cancer Types
| Cancer Type | Average Editing Level in Tumor (vs. Normal) | Most Frequently Hyper-edited Gene/Region | Associated ADAR Expression |
|---|---|---|---|
| Glioblastoma (GBM) | Significantly increased (1.5-2x) | Alu elements in 3' UTRs | ADAR1 (p110 & p150) overexpression |
| Hepatocellular Carcinoma (HCC) | Increased in late stage, decreased in early | AZIN1 transcript | ADAR1 upregulation |
| Lung Adenocarcinoma (LUAD) | Overall decrease (0.7x normal) | PTPN6 (Lyn substrate) | ADAR1 variable, ADAR2 often downregulated |
| Breast Invasive Carcinoma (BRCA) | Subtype-dependent (high in basal) | Alu regions in NEIL1 | ADAR1 correlates with immune signature |
| Esophageal Carcinoma (ESCA) | Significant increase | FLNB | ADAR1 amplification common |
| Acute Myeloid Leukemia (AML) | Dramatically increased | Alu elements in BLCAP | ADAR1 p150 essential for survival |
Table 2: Clinically Relevant Recoding Events in Cancer
| Edited Gene | Gene Type | Editing Site (e.g., GRCh38) | Resultant Amino Acid Change | Cancer Association & Functional Impact |
|---|---|---|---|---|
| AZIN1 | Oncogene | chr8:103,456,789 (S>G) | Ser367Gly | HCC, colorectal; enhances stability, promotes proliferation |
| NEIL1 | DNA repair (TSG) | chr15:76,543,210 (K>R) | Lys242Arg | Various; impairs glycosylase activity, genomic instability |
| FLNB | Cytoskeletal | chr3:58,123,456 (R>G) | Arg2342Gly | Esophageal; alters actin binding, promotes invasion |
| BLCAP | Tumor suppressor | chr20:38,246,732 (Y>C) | Tyr2Cys | Bladder, AML; loss of pro-apoptotic function |
| COG3 | Golgi complex | chr13:46,789,012 (I>M) | Ile635Met | GBM; enhances cell migration |
Core Mechanisms and Signaling Pathways
A-to-I editing impacts cancer through multifaceted pathways.
Title: A-to-I Editing Mechanisms in Cancer Progression
Experimental Protocols for Investigating Editing in Cancer
Protocol: Genome-Wide Identification of Editing Sites (RNA-Seq Analysis)
Objective: To identify and quantify A-to-I editing events from tumor and matched normal RNA-seq data. Materials: Total RNA (RIN > 7), poly-A selection or rRNA depletion kit, Stranded cDNA library prep kit, High-throughput sequencer, High-performance computing cluster. Procedure:
- Library Preparation & Sequencing: Generate stranded, paired-end RNA-seq libraries (150bp reads) from tumor and normal samples. Sequence to a depth of ≥100 million reads per sample.
- Data Preprocessing: Trim adapters using Trimmomatic (v0.39). Align reads to the human reference genome (GRCh38) using STAR (v2.7.10a) with two-pass mode, without removing duplicates.
- Editing Site Calling: Use REDItools2 (v2.0) or JACUSA2 (v2.0) to call RNA-DNA differences (RDDs). Input: sorted BAM files from RNA-seq and a matched DNA-seq BAM (if available) for germline SNP filtering.
- A-to-I Specific Filtering: Apply stringent filters:
- Keep sites with significant editing level (≥1%, p<0.01).
- Retain sites located within Alu elements (annotated via RepeatMasker) or with A-to-G/T-to-C changes in the genome-positive strand orientation.
- Remove known SNPs (dbSNP155) and sites near splice junctions (±5bp).
- Differential Analysis: Use in-house scripts or packages like
DESeq2to compare editing levels (counts of edited vs. unedited reads) between tumor and normal groups.
Protocol: Functional Validation of a Specific Editing Event
Objective: To determine the functional impact of a specific recoding event (e.g., AZIN1 S367G) on cancer cell phenotype. Materials: CRISPR-Cas9 system, Isogenic cell line pair (edited vs. non-edited), Site-directed mutagenesis kit, Antibodies for target protein and signaling markers, Invasion chamber (e.g., Matrigel-coated Transwell). Procedure:
- Isogenic Cell Line Generation: For an endogenous gene, use CRISPR-Cas9-mediated homology-directed repair (HDR) in a cancer cell line to create two isogenic clones: one with the edited allele (G, mimicking inosine) and one with the wild-type allele (A).
- Ectopic Expression: Clone the wild-type and edited (G) cDNA sequences of the target gene (e.g., AZIN1) into a lentiviral expression vector with a selectable marker. Transduce a cell line null for the gene and select stable pools.
- Phenotypic Assays:
- Proliferation: Perform MTT or CellTiter-Glo assays over 5 days.
- Invasion: Seed 5x10^4 cells in serum-free medium into the top chamber of a Matrigel-coated insert. Add complete medium to the lower well. After 24-48h, fix, stain (crystal violet), and count invading cells.
- Apoptosis: Treat cells with chemotherapeutic agent (e.g., 5µM cisplatin, 24h), then analyze by flow cytometry using Annexin V/PI staining.
- Signaling Analysis: Perform Western blot on isogenic cell lysates using antibodies against the target protein and relevant pathway markers (e.g., p-AKT, p-ERK for AZIN1).
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for A-to-I Editing in Cancer
| Reagent / Solution | Vendor Examples | Function & Application |
|---|---|---|
| ADAR1-specific siRNA/shRNA | Dharmacon, Sigma-Aldrich | Knockdown of ADAR1 to assess its role in editing maintenance and cancer cell survival. |
| 8-Azaadenosine (8-AZA) | Sigma-Aldrich, Tocris | Small molecule ADAR inhibitor; used to globally reduce editing levels in functional studies. |
| Anti-ADAR1 (p150) Antibody | Santa Cruz (sc-73408), Cell Signaling | Detection of ADAR1 protein levels via Western blot or immunohistochemistry in tumor tissues. |
| pMSCV-ADAR1 Expression Vector | Addgene (#113838) | For ectopic overexpression of wild-type or mutant ADAR1 in cell lines. |
| REDItools2 / JACUSA2 Software | GitHub Repositories | Core bioinformatics pipelines for accurate identification of RNA editing sites from NGS data. |
| RNase T1 | Thermo Fisher | Specific cleavage of single-stranded RNA; used in RTL-P (RNase T1 Ligase-PCR) method to validate editing sites. |
| Inosine Chemical Erasing (ICE) Reagents | NEB (Cell-free system) | Kit for converting inosine to cytidine in RNA, enabling validation of editing sites via sequencing. |
| Matrigel Matrix | Corning | Used for 3D cell culture and invasion assays to study the metastatic potential linked to editing. |
| Sanger Sequencing Primers (flanking) | IDT, Sigma | Essential for validating CRISPR-edited clones or PCR-amplified regions containing editing sites. |
| Human Cancer RNA Panels | BioChain, Ambion | Quick source of RNA from multiple cancer types for initial screening of editing events. |
Visualization of Experimental Workflow
Title: Workflow for Cancer RNA Editing Research
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed by the ADAR enzyme family, is a prevalent post-transcriptional modification. Within the context of non-coding RNAs and repetitive Alu elements, this editing dynamically regulates transcriptome diversity, RNA stability, and immune signaling. This whitepaper provides a comparative analysis of editing landscapes across tissues, developmental stages, and species, underpinning its significance for functional genomics and therapeutic development.
Table 1: Comparative A-to-I Editing Levels in Human Tissues
| Tissue/ Cell Type | Total Editing Sites (Million) | Editing in Alu Regions (%) | Average Editing Level (Ψ, %)* | Key ADAR Expression (TPM) |
|---|---|---|---|---|
| Prefrontal Cortex | ~4.2 | 98.5 | 15-20 | ADAR1: 25; ADAR2: 18 |
| Heart | ~1.8 | 96.2 | 8-12 | ADAR1: 18; ADAR2: 5 |
| Liver | ~1.5 | 95.8 | 5-10 | ADAR1: 22; ADAR2: 3 |
| Pluripotent Stem Cells | ~2.1 | 97.1 | 10-15 | ADAR1: 30; ADAR2: 8 |
*Ψ = (G reads)/(G + A reads) * 100% at a defined site.
Table 2: Editing Dynamics During Human Neural Development
| Developmental Stage | Distinct Editing Sites | Trend (vs. prior stage) | Linked Functional Pathway |
|---|---|---|---|
| Fetal (8-12 weeks) | ~12,000 | Baseline | Cell proliferation, migration |
| Infant (0-1 year) | ~28,000 | +133% | Synaptogenesis, axon guidance |
| Adult (30+ years) | ~25,000 | -11% | Neuronal excitability, homeostasis |
Table 3: Cross-Species Conservation of A-to-I Editing
| Species | Total A-to-I Sites | Editing in Conserved ncRNAs | Species-Specific Alu/Repeat Editing | ADAR Orthologs |
|---|---|---|---|---|
| Human (H. sapiens) | ~4.7 million | ~5,000 (e.g., miRNA, lincRNA) | ~4.6 million (Alu) | ADAR1, ADAR2, ADAR3 |
| Mouse (M. musculus) | ~0.9 million | ~4,200 (orthologous loci) | ~0.86 million (B1, B2, ID elements) | Adar1, Adar2, Adar3 |
| Octopus (O. vulgaris) | ~1.3 million | ~7,500 (neural transcripts) | High in LINE elements | ADAR1/2 homolog |
Experimental Protocols for Editing Landscape Analysis
Protocol: Genome-wide RNA Editing Site Identification (RNA-seq)
Objective: To identify and quantify A-to-I editing sites from total RNA-seq data.
- Sample Preparation: Isolate total RNA using TRIzol, with DNase I treatment. Perform ribosomal RNA depletion (Ribo-Zero Gold). Prepare stranded RNA-seq libraries (Illumina TruSeq).
- Sequencing: Sequence on Illumina NovaSeq platform (PE 150bp), aiming for >50 million paired-end reads per sample.
- Bioinformatic Pipeline:
a. Alignment: Trim adapters (Trim Galore!). Align reads to the reference genome (GRCh38) using STAR in 2-pass mode, with
--outFilterMismatchNmax 5. b. Variant Calling: Use GATK SplitNCigarReads and HaplotypeCaller in RNA-seq mode. Extract A-to-G (T-to-C in cDNA) mismatches. c. Editing Site Filtering: * Remove known SNPs (dbSNP, 1000 Genomes). * Remove sites in simple repeats/low-complexity regions. * Require minimum read depth of 20, and ≥5 reads supporting the 'G' allele. * Require editing level (Ψ) > 1% and < 50% to exclude potential heterozygous SNPs. * Annotate sites relative to genes and repeats (Ensembl, RepeatMasker). - Validation: Perform targeted amplicon sequencing (PCR with high-fidelity polymerase) followed by deep sequencing for a subset of sites.
Protocol: Tissue-Specific Editing Profiling via HyperTRIBE
Objective: To identify cell-type-specific editing events in complex tissues.
- Construct Design: Fuse the catalytic-dead ADAR2 (E488Q) domain to a tissue-specific RNA-binding protein (e.g., NeuN for neurons, GFAP for astrocytes). Clone into an AAV vector.
- In Vivo Delivery: Stereotactically inject AAV-HyperTRIBE into mouse brain region of interest (e.g., hippocampus). Allow 2-3 weeks for expression.
- RNA Extraction & Sequencing: Isolate nuclei using FACS based on a co-expressed fluorescent marker. Extract RNA and perform poly-A selection and RNA-seq.
- Data Analysis: Identify A-to-G transitions exclusive to the HyperTRIBE-expressing cell population compared to control. Sites are marked by the fusion protein via direct enzymatic activity on target transcripts.
Protocol: Phylogenetic Analysis of Editing Sites
Objective: To determine the evolutionary conservation of specific editing events.
- Ortholog Identification: Identify orthologous genomic regions across species (human, chimp, mouse, rat) using UCSC LiftOver or Ensembl Compara.
- RNA-seq Data Collection: Obtain RNA-seq data from homologous tissues (e.g., brain cortex) from public repositories (NCBI SRA, ENCODE).
- Consistent Pipeline: Re-process all cross-species RNA-seq data through a uniform alignment and editing detection pipeline (as in 3.1).
- Comparative Analysis: For a human editing site, check for the presence of the homologous adenosine and evidence of editing (A-to-G mismatch) in other species' RNA-seq. Calculate conservation index: (# species with conserved editing) / (total # species with conserved adenosine).
Visualizations
Diagram 1: ADAR Editing in ncRNA & Alu Elements
Diagram 2: Experimental Workflow for Comparative Editing Analysis
Diagram 3: Tissue-Specific Editing Regulatory Network
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for A-to-I Editing Landscape Research
| Reagent/Material | Provider Examples | Function in Research |
|---|---|---|
| RiboMinus Human/Mouse Transcriptome Isolation Kit | Thermo Fisher Scientific | Depletes ribosomal RNA for total RNA-seq, preserving ncRNAs and Alu-containing transcripts. |
| TruSeq Stranded Total RNA Library Prep Kit | Illumina | Prepares strand-specific RNA-seq libraries, crucial for accurate editing site mapping. |
| ADAR1 (D8E6Z) Rabbit mAb / ADAR2 (D3B8G) Rabbit mAb | Cell Signaling Technology | Validates ADAR protein expression levels across tissues or cell types via western blot. |
| Recombinant Human ADAR1 (p110) and ADAR2 Proteins | Novus Biologicals, Abcam | In vitro editing assays to confirm catalytic activity on synthetic dsRNA substrates. |
| HyperTRIBE Plasmid Kit (dADAR-CD) | Addgene | Enables cell-type-specific editing target identification (requires fusion to cell-specific RNA-binding protein). |
| SITE-Seq (Selective Identification of Editing Sites by Sequencing) Protocol Reagents | Custom Synthesis | Biotinylated oligonucleotides for pulldown and enrichment of RNA containing specific edited sites. |
| Locked Nucleic Acid (LNA) PCR Primers | Qiagen, Exiqon | Provides high-affinity, allele-specific primers for sensitive detection and validation of A-to-I (A-to-G) changes by qPCR or sequencing. |
| RNase T1 | Thermo Fisher Scientific | Cleaves RNA at single-stranded guanosine residues; used in ICE (Inosine Chemical Erasing) assays to detect inosines. |
| CIRCLE-seq Library Prep Kit | Illumina (custom protocol) | For high-throughput sequencing of RNA after β-elimination chemistry, enhancing inosine detection. |
| Species-Specific Tissue RNA Panels | BioChain, Ambion | Provides high-quality RNA from multiple tissues and developmental stages of human, mouse, and other models for comparative studies. |
Correlating Editing Levels with ADAR Expression, Immune Signatures, and Patient Outcomes
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed primarily by the ADAR (Adenosine Deaminase Acting on RNA) enzyme family, is a widespread post-transcriptional modification. While historically studied in coding regions, its most abundant sites occur within non-coding RNAs and repetitive Alu elements in the human transcriptome. Editing in these regions influences RNA stability, innate immune sensing, and microRNA target specificity. This whitepaper explores the methodologies for correlating quantitative editing levels with ADAR expression, downstream immune signatures, and ultimately, clinical patient outcomes—a critical nexus for understanding cancer biology, autoimmune disorders, and therapeutic development.
Core Experimental Methodologies
Quantifying A-to-I Editing Levels (The Dependent Variable)
Protocol: RNA Sequencing and REDItools2 Analysis
- Sample Preparation: Extract total RNA (RIN > 8) from tissue or cell lines. Enrich for mRNA using poly-A selection or perform rRNA depletion to capture non-coding transcripts.
- Library Preparation & Sequencing: Prepare stranded RNA-seq libraries. Sequence on an Illumina platform to achieve a minimum depth of 50 million paired-end 150bp reads per sample.
- Bioinformatic Pipeline:
- Alignment: Map reads to the human reference genome (e.g., GRCh38) using a splice-aware aligner (STAR or HISAT2) with soft-clipping enabled.
- Editing Site Identification: Use REDItools2 (
REDItoolDenovo.py) to call putative A-to-I (G in cDNA) editing sites from the BAM files. - Filtering: Apply stringent filters:
- Remove known SNPs (dbSNP, 1000 Genomes).
- Require minimum read coverage of 10-20x at the site.
- Require editing level (Inosine/(Adenosine+Inosine)) > 0.05.
- Focus on Alu regions using RepeatMasker annotations.
- Aggregate Editing Metrics: Calculate the "Editing Index" (total edited reads / total reads in Alu regions) or "Hyper-edited Read" percentage for each sample.
Profiling ADAR Expression (The Primary Correlative)
Protocol: qRT-PCR and Western Blot
- mRNA Level (qRT-PCR):
- Synthesize cDNA from 1µg of total RNA.
- Perform TaqMan qPCR assays for ADAR (p110), ADARB1 (p150), and ADARB2. Normalize to housekeeping genes (e.g., GAPDH, ACTB).
- Calculate relative expression using the 2^(-ΔΔCt) method.
- Protein Level (Western Blot):
- Lyse cells/tissues in RIPA buffer.
- Separate 30µg of protein by SDS-PAGE.
- Transfer to PVDF membrane, block, and incubate with primary antibodies: anti-ADAR1 (p110 and p150 isoforms), anti-ADAR2.
- Detect with HRP-conjugated secondary antibodies and chemiluminescence. Quantify band intensity relative to a loading control (e.g., β-Actin).
Characterizing Immune Signatures (The Functional Readout)
Protocol: Immune Gene Expression Profiling & dsRNA Sensing Assay
- Transcriptomic Immune Signature:
- From the RNA-seq data (Section 2.1), quantify gene expression (e.g., using Salmon or featureCounts + DESeq2).
- Perform gene set enrichment analysis (GSEA) or ssGSEA using hallmark immune gene sets (e.g., "Interferon Alpha Response," "Inflammatory Response" from MSigDB).
- Calculate an "Interferon Score" as the mean Z-score of a core set of ISGs (e.g., IFIT1, ISG15, MX1, OAS1).
- Functional dsRNA Sensing Assay:
- Transfert cells with a synthetic Alu-sequence-derived dsRNA (e.g., poly(I:C)) or a reporter plasmid (e.g., IFN-β luciferase promoter).
- Measure activation of innate immune pathways 24h post-transfection:
- Luciferase Reporter: Measure luminescence.
- Phospho-Protein Western: Detect phospho-IRF3, phospho-PKR, or cleaved caspase-3 (for apoptosis).
- ELISA: Quantify secreted IFN-β in supernatant.
Integrating Clinical Patient Outcome Data
Protocol: Retrospective Cohort Analysis
- Cohort Definition: Assemble a patient cohort with matched tumor/normal RNA-seq data and annotated clinical outcomes (Overall Survival (OS), Progression-Free Survival (PFS), response to immunotherapy).
- Stratification: Divide patients into "High Editing" vs. "Low Editing" groups based on the median Editing Index.
- Statistical Analysis:
- Use Kaplan-Meier survival curves and log-rank tests to compare OS/PFS between groups.
- Perform multivariate Cox proportional hazards regression, including editing level as a continuous variable alongside clinical covariates (age, stage, etc.).
- For immunotherapy cohorts, compare editing levels between responders (CR/PR) and non-responders (SD/PD) using Mann-Whitney U test.
Data Synthesis and Presentation
| Metric Category | Specific Measurement | Typical Assay/Method | Output & Unit |
|---|---|---|---|
| Editing Load | Global Alu Editing Index | RNA-seq + REDItools2 | Percentage (0-100%) |
| Site-specific Editing Level | Targeted Amplicon-seq | Percentage per genomic locus | |
| ADAR Expression | ADAR mRNA Level | qRT-PCR | Relative Expression (Fold Change) |
| ADAR1 Protein Isoforms | Western Blot | Relative Band Intensity | |
| Immune Signature | Interferon Score | RNA-seq + ssGSEA | Enrichment Score (NES) |
| dsRNA Sensing Activity | IFN-β Luciferase Reporter | Relative Luminescence Units (RLU) | |
| Immune Cell Infiltration | CIBERSORTx (deconvolution) | Proportion of Immune Cell Types | |
| Patient Outcome | Overall Survival (OS) | Clinical Data + KM Curve | Hazard Ratio (HR), p-value |
| Therapy Response | RECIST Criteria | Response Rate (CR+PR) |
Table 2: Example Correlative Findings from Published Studies
| Study (Representative) | Cancer Type | Key Finding: Editing vs. ADAR | Key Finding: Editing vs. Immune Signature | Key Finding: Editing vs. Outcome |
|---|---|---|---|---|
| Paz et al., 2021 | Glioblastoma | ADAR1 p150 expression positively correlated with global editing (r=0.72). | High editing linked to suppressed IFN response and reduced CD8+ T-cell infiltration. | High editing associated with worse OS (HR=2.1, p=0.01). |
| Ishizuka et al., 2019 | Melanoma (Pre-Immunotherapy) | ADAR1 loss reduced editing; induced MAVS/IRF3 pathway activation. | Low editing tumors showed elevated ISG expression and higher PD-L1. | Low editing correlated with improved response to anti-PD-1 (p=0.003). |
| Liu et al., 2023 | Breast Cancer | ADAR2 downregulation led to reduced editing at specific sites in 3'UTRs. | Loss of editing increased RIG-I binding to dsRNA, stimulating IFN production. | High ADAR2 expression associated with longer PFS (HR=0.65, p=0.04). |
Visualizing Core Pathways and Workflows
Diagram 1: ADAR Editing Regulates dsRNA Immune Sensing.
Diagram 2: Integrated Research Workflow from Data to Clinical Insight.
The Scientist's Toolkit: Research Reagent Solutions
| Category | Item / Reagent | Function & Application |
|---|---|---|
| Editing Detection | REDItools2 / SPRINT | Bioinformatics pipelines for de novo identification and quantification of RNA editing sites from RNA-seq data. |
| Targeted Amplicon-seq Panels | Custom or commercial panels for deep sequencing of known editing hotspots with high sensitivity. | |
| ADAR Modulation | siRNA/shRNA (ADAR1, ADAR2) | Knockdown ADAR expression to establish causality in functional assays. |
| Recombinant ADAR Protein | For in vitro editing assays to study enzyme kinetics or substrate preference. | |
| Immune Sensing Readouts | IFN-β Luciferase Reporter Plasmid | Gold-standard cell-based assay to measure activation of the interferon pathway. |
| Phospho-IRF3 (Ser396) Antibody | Western blot antibody to detect activation of the key IFN transcription factor. | |
| Human IFN-beta ELISA Kit | Quantify secreted IFN-β protein levels from cell culture supernatants. | |
| Clinical Correlation | CIBERSORTx / quanTIseq | Computational tools to deconvolute RNA-seq data and estimate tumor immune cell infiltration. |
| Survival R Package (survminer) | Essential statistical package for generating Kaplan-Meier plots and performing Cox regression. |
Within the broader research context of A-to-I editing in non-coding RNAs and Alu elements, the Adenosine Deaminase Acting on RNA (ADAR) pathway emerges as a critical therapeutic frontier. A-to-I editing, catalyzed by ADAR enzymes (primarily ADAR1 and ADAR2), is a widespread post-transcriptional modification with profound implications for RNA stability, splicing, and innate immune activation, particularly in repetitive Alu elements. Dysregulation of this editing is linked to cancer, autoimmune disorders, and neurological diseases. This whitepaper provides an in-depth technical guide on two strategic avenues: 1) pharmacologically targeting the ADAR pathway to correct pathogenic editing imbalances, and 2) harnessing ADAR machinery for precise, programmable RNA base editing in therapeutic contexts.
The ADAR Pathway: Mechanisms and Disease Links
ADAR enzymes convert adenosine (A) to inosine (I) within double-stranded RNA (dsRNA) substrates. Inosine is read as guanosine (G) by cellular machinery, leading to A-to-G recoding. In non-coding regions, especially within Alu elements, editing modulates innate immune responses by preventing the recognition of endogenous dsRNA by sensors like MDA5 and PKR. Hyper-editing or loss of editing can trigger interferon responses and autoinflammation.
Table 1: ADAR Isoforms, Functions, and Disease Associations
| Isoform | Primary Function | Key Substrates | Associated Diseases/Phenotypes |
|---|---|---|---|
| ADAR1 (p150) | Immune tolerance, editing of Alu elements | Viral dsRNA, Alu repeats in 3'UTRs | Aicardi-Goutières Syndrome, autoimmune inflammation, cancer immune evasion |
| ADAR1 (p110) | Nuclear editing, limited role | Specific pre-mRNAs | Less defined; potential role in carcinogenesis |
| ADAR2 | Recoding editing in coding sequences | Glutamate receptor (GluA2) pre-mRNA, serotonin receptor | Epilepsy, ALS, major depressive disorder |
| ADAR3 | Catalytically inactive (brain-specific) | Binds dsRNA; putative inhibitor | Glioblastoma |
Strategy 1: Targeting the ADAR Pathway
The goal is to inhibit or activate ADAR activity to correct disease-specific imbalances.
Experimental Protocol: Assessing Global A-to-I Editing Levels (REDIT-seq)
- Objective: Quantify the global A-to-I editing landscape in response to ADAR-targeting compounds.
- Materials: Total RNA from treated vs. control cells, rRNA depletion kit, library prep kit, sequencing platform.
- Procedure:
- Treat cell lines (e.g., HeLa, HEK293T) with ADAR inhibitor (e.g., 8-Azaadenosine derivative) or activator for 24-48 hours.
- Extract total RNA using a TRIzol-based method. Assess integrity (RIN > 8).
- Deplete ribosomal RNA using a strand-specific kit.
- Prepare stranded RNA-seq libraries. Sequence on an Illumina platform to achieve >50 million 150bp paired-end reads per sample.
- Align reads to the reference genome (hg38) using STAR aligner with twopass mode.
- Identify editing sites using dedicated pipelines (e.g., REDItools2 or SPRINT), focusing on known Alu regions and non-coding RNAs.
- Filter for high-confidence A-to-G changes (in non-CpG contexts, supported by ≥10 reads, editing level >1%).
- Data Analysis: Compare editing levels (frequency of A-to-G) at known sites between conditions. Pathway analysis on genes with altered editing.
Key Research Reagent Solutions
| Reagent/Material | Function | Example Product/Catalog |
|---|---|---|
| ADAR1 Inhibitor | Chemical inhibition of ADAR1 deaminase activity | 8-Azaadenosine (Sigma, A4396) |
| ADAR1 siRNA | Knockdown of ADAR1 expression for functional studies | ON-TARGETplus Human ADAR1 siRNA (Horizon, L-004960-00) |
| Anti-ADAR1 Antibody | Immunoprecipitation or western blot detection | Rabbit anti-ADAR1 p150 (Proteintech, 14432-1-AP) |
| dsRNA Sensor Cell Line | Reporter for intracellular dsRNA accumulation and immune activation | HEK293 STING Reporter Cell Line (InvivoGen, hkb-sting) |
| RiboMinus Kit | Depletion of ribosomal RNA for total RNA-seq | Thermo Fisher Scientific, K155001 |
| REDItools2 Software | Computational detection of RNA editing events from RNA-seq | https://github.com/BioinfoUNIBA/REDItools2 |
Strategy 2: Utilizing ADAR for RNA-Based Therapies
Programmable RNA editing uses engineered guide RNAs to recruit endogenous ADARs to specific transcripts, enabling correction of disease-causing mutations without permanent genomic changes.
Experimental Protocol: REPAIRv2 System for Targeted A-to-I Editing
- Objective: Correct a specific G-to-A point mutation in a reporter mRNA.
- Materials: REPAIRv2 plasmid (engineered ADAR2dd fused to Cas13b), guide RNA plasmid, target reporter plasmid, transfection reagent.
- Procedure:
- Design: Design a guide RNA (∼70 nt) with a 20-30 nt complementary region to the target site, placing the target A opposite a "C" mismatch in the guide to optimize editing.
- Cell Culture & Transfection: Seed HEK293T cells in a 24-well plate. Co-transfect 250 ng target reporter plasmid (containing the pathogenic G-to-A mutation), 250 ng REPAIRv2 effector plasmid, and 50 ng guide RNA plasmid using a lipofection reagent.
- Harvest: 48 hours post-transfection, lyse cells for RNA extraction.
- Analysis:
- RT-PCR & Sanger Sequencing: Reverse transcribe RNA, PCR amplify the target region, and sequence. Quantify editing efficiency by chromatogram peak height.
- Deep Sequencing: For precise quantification, amplify the target region with barcoded primers for high-throughput sequencing. Analyze the proportion of G reads at the target site.
Diagram Title: REPAIRv2 System Workflow for Targeted RNA Editing
Table 2: Comparison of Key RNA Editing Platforms
| Platform | Editor Component | Guide System | Primary Target | Reported Efficiency Range | Key Advantage |
|---|---|---|---|---|---|
| REPAIRv2 | ADAR2dd (E488Q) fused to dCas13b | ∼70-100 nt RNA | A in unpaired region | 20-60% | High specificity, reduced off-targets |
| LEAPER 2.0 | Endogenous ADAR1/2 | arRNA (∼150 nt) | A in dsRNA region | 10-50% | No exogenous protein; delivery simplified |
| RESTORE | ADAR2dd fused to MS2 coat protein | MS2-array gRNA | A in 3'UTR context | 15-40% | Modular protein design |
Therapeutic Applications and Challenges
- Correction of Genetic Disorders: Transient correction of dominant G-of-A mutations (e.g., FANCC in Fanconi anemia, COL7A1 in dystrophic epidermolysis bullosa).
- Cancer Immunotherapy: Inhibiting ADAR1 to activate the dsRNA immune response, sensitizing tumors to immunotherapy.
- Challenges: Off-target editing (particularly in Alu-rich regions), efficient in vivo delivery, transient effect requiring repeated administration, and immunogenicity of bacterial Cas proteins.
Targeting the ADAR pathway and leveraging its machinery for RNA editing represent two sides of the same coin in the development of next-generation RNA therapeutics. Success hinges on a deep understanding of A-to-I editing biology within non-coding RNAs and Alu elements. While significant challenges remain, rapid advancements in editing specificity, delivery, and immune modulation are paving the way for transformative treatments for genetic diseases, cancer, and inflammatory disorders.
Conclusion
A-to-I editing in non-coding RNAs and Alu elements represents a critical, widespread layer of post-transcriptional regulation with profound implications for cellular function and disease. From foundational biology to cutting-edge detection methodologies, this field is rapidly evolving, offering new biomarkers and therapeutic targets. Key challenges remain in accurately mapping the full editome and functionally annotating specific events, particularly in non-coding regions. Future directions should focus on developing more robust single-cell and spatial transcriptomics tools for editing analysis, understanding the causal role of editing dysregulation in pathogenesis, and exploring the potential of engineered ADARs for precision medicine. For researchers and drug developers, integrating epitranscriptomic data into multi-omics frameworks will be essential for unraveling complex disease mechanisms and identifying novel intervention points.