Bulk RNA Sequencing: A Comprehensive Guide from Principles to Clinical Applications
This article provides a complete overview of bulk RNA sequencing, covering its foundational principles, step-by-step methodological workflow, and diverse applications in biomedical research and drug development.
Bulk RNA Sequencing: A Comprehensive Guide from Principles to Clinical Applications
Abstract
This article provides a complete overview of bulk RNA sequencing, covering its foundational principles, step-by-step methodological workflow, and diverse applications in biomedical research and drug development. It details the experimental process from sample preparation to bioinformatics analysis, addresses key troubleshooting and optimization strategies for robust results, and offers a comparative analysis with emerging single-cell technologies. Aimed at researchers and drug development professionals, this guide synthesizes current best practices and future directions, empowering readers to effectively design, implement, and interpret bulk RNA-seq studies for advancing personalized medicine and clinical diagnostics.
The Essential Guide to Bulk RNA-Seq Principles and Technology
The journey of transcriptomics has been marked by a series of revolutionary technological advances, with RNA sequencing (RNA-seq) representing one of the most significant breakthroughs in functional genomics. This evolution from Sanger sequencing to next-generation sequencing (NGS) has fundamentally transformed how researchers investigate gene expression, discover novel transcripts, and understand cellular mechanisms. For researchers focused on bulk RNA sequencing—which measures the average gene expression across populations of thousands to millions of cells—this technological progression has enabled increasingly sophisticated investigations into development, disease mechanisms, and drug responses. This technical guide examines the key transitions in RNA sequencing technologies, their impact on bulk RNA-seq methodologies, and the practical considerations for contemporary research applications.
Historical Context: From Sanger to Sequencing Revolution
The foundation of DNA sequencing was established in the 1970s with Frederick Sanger's development of the chain-termination method, also known as dideoxy sequencing [1]. This approach, which became known as Sanger sequencing, relied on fluorescently-labeled dideoxynucleotides (ddNTPs) that terminate DNA strand elongation at specific nucleotide positions, followed by capillary gel electrophoresis to separate the fragments by size and determine the sequence [1] [2].
Sanger sequencing provided the foundation for transcriptomics through expressed sequence tag (EST) libraries and sequencing of cDNA clones [3]. However, these approaches were relatively low throughput, expensive, and generally not quantitative [3]. Tag-based methods like serial analysis of gene expression (SAGE) and massively parallel signature sequencing (MPSS) were developed to overcome some limitations but still faced challenges with mapping short tags and distinguishing isoforms [3].
The critical limitation of Sanger sequencing was its fundamental throughput constraint—it could only sequence a single DNA fragment at a time [4]. This bottleneck made comprehensive transcriptome analysis prohibitively expensive and time-consuming, setting the stage for a paradigm shift with the arrival of NGS technologies.
The Next-Generation Sequencing Revolution
Next-generation sequencing technologies transformed transcriptomics by introducing massively parallel sequencing, enabling millions of DNA fragments to be sequenced simultaneously in a single run [4]. This fundamental shift in scale provided several decisive advantages over Sanger sequencing for RNA analysis, particularly for bulk RNA-seq applications.
Table 1: Key Technical Comparisons Between Sanger Sequencing and Next-Generation Sequencing
| Aspect | Sanger Sequencing | Next-Generation Sequencing (NGS) |
|---|---|---|
| Throughput | Low (one fragment at a time) [4] | High (millions of fragments simultaneously) [4] |
| Detection Limit | ~15-20% [4] [1] | As low as 1% for low-frequency variants [4] [2] |
| Discovery Power | Limited for novel variants [4] | High discovery power for novel transcripts/isoforms [4] [5] |
| Dynamic Range | Limited | >8,000-fold [3] |
| Read Length | 500-700 bp [1] | 30-400 bp (technology-dependent) [3] |
| Cost Effectiveness | Good for 1-20 targets [4] | Better for larger numbers of targets/samples [4] |
The advantages of NGS for transcriptome analysis are profound. RNA-seq provides a "far more precise measurement of levels of transcripts and their isoforms than other methods" with a dynamic range spanning over 8,000-fold [3]. Unlike hybridization-based approaches like microarrays, RNA-seq is not limited to detecting transcripts that correspond to existing genomic sequence, has minimal background signal, and requires less RNA sample [3]. These characteristics make it particularly valuable for discovering novel transcripts, alternative splice sites, and gene fusions [5].
Bulk RNA-Sequencing: Methodology and Workflow
Bulk RNA-seq is a widely used technique that measures gene expression in samples consisting of large pools of cells, such as tissues, whole organs, or sorted cell populations [6] [7] [8]. This approach provides a population-level average gene expression profile, making it valuable for differential expression analysis between conditions (e.g., diseased vs. healthy, treated vs. control) and for obtaining global expression profiles from whole tissues or organs [8].
Experimental Workflow
The standard bulk RNA-seq workflow involves multiple critical steps:
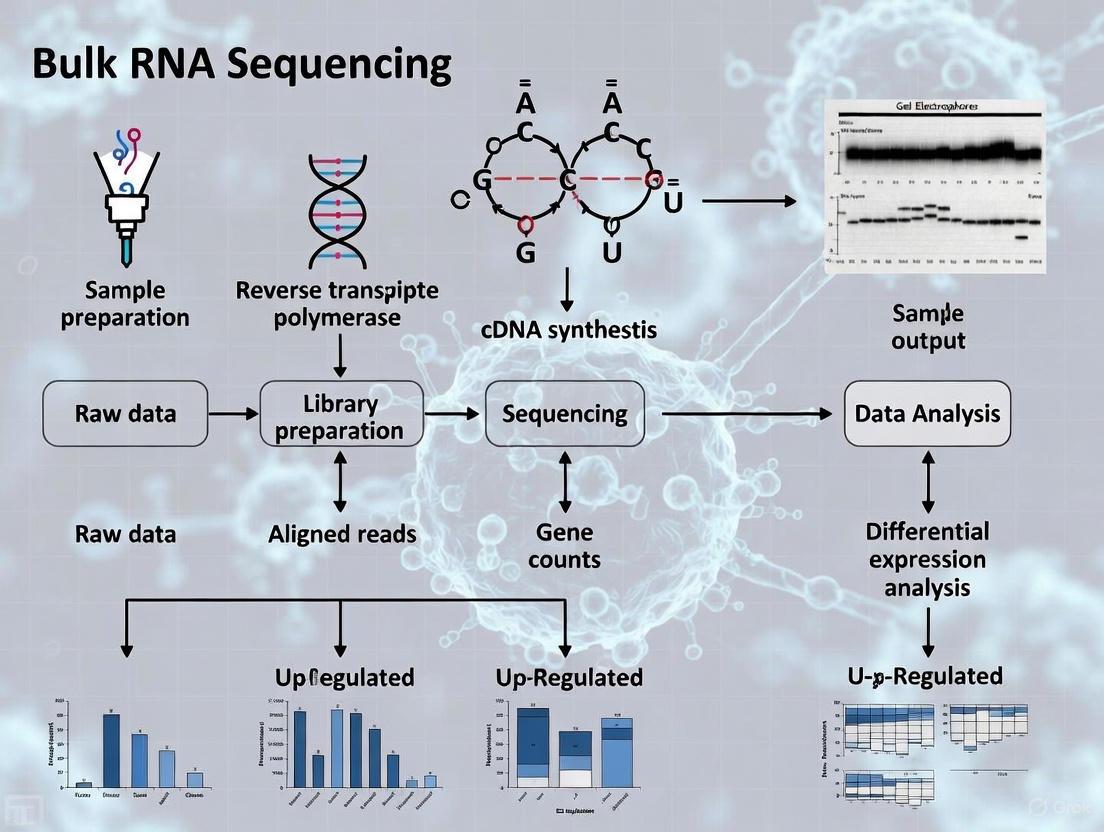
Sample Preparation and RNA Extraction: Biological samples are processed to extract total RNA. Due to ribosomal RNA (rRNA) typically constituting 80-95% of the total RNA population, it is usually removed to focus sequencing on transcripts of interest [5]. This is achieved through either poly(A) selection to enrich for messenger RNA (mRNA) or ribosomal depletion [5] [7].
Library Preparation: The RNA is converted to complementary DNA (cDNA) through reverse transcription, since current NGS platforms sequence DNA rather than RNA directly. The cDNA is then fragmented into appropriate sizes (typically 200-500 bp) for sequencing, and adapters are ligated to the fragments [3] [9]. These adapters facilitate the sequencing reaction and often include barcodes to allow multiplexing of multiple samples.
Sequencing: The prepared libraries are loaded onto NGS platforms where massive parallel sequencing occurs, generating millions of short reads [3]. The specific read length (30-400 bp) and number of reads per sample depend on the sequencing technology and experimental design [3].
Data Analysis: The generated sequences (raw reads) undergo a computational pipeline including quality control, alignment to a reference genome/transcriptome, and quantification of gene expression levels [10] [9].
Diagram 1: Bulk RNA-seq experimental workflow
Quantification and Bioinformatics Analysis
A critical challenge in RNA-seq is converting raw sequencing data into accurate gene expression measurements. This involves addressing two levels of uncertainty: "identifying the most likely transcript of origin of each RNA-seq read" and "converting read assignments to a count matrix" that models the uncertainty inherent in many read assignments [6].
Two primary computational approaches have emerged for this quantification:
Alignment-Based Methods: Tools like STAR or HISAT2 perform formal alignment of sequencing reads to either a genome or transcriptome, producing detailed alignment maps that record exact coordinates of sequence matches [6] [9]. The aligned reads are then assigned to genes using tools like featureCounts [9].
Pseudoalignment Methods: Tools such as Salmon and kallisto use lightweight algorithms that perform probabilistic mapping of reads to transcripts without exact base-level alignment, significantly improving speed [6]. These tools simultaneously address both levels of uncertainty—read origin and count conversion—and are particularly valuable for large-scale studies.
For bulk RNA-seq analysis, the end result is typically a count matrix where rows represent genes and columns represent samples, with each cell containing the estimated number of reads originating from that gene in that sample [6]. This matrix serves as the input for downstream differential expression analysis using statistical methods in tools like limma or DESeq2 [6] [10].
Diagram 2: Bulk RNA-seq data analysis pipeline
Advanced Methodological Considerations
Strand-Specific Libraries
An important methodological consideration is the construction of strand-specific libraries, which preserve information about the transcriptional orientation of reads [3]. This is particularly valuable for transcriptome annotation, especially in genomic regions with overlapping transcription from opposite strands [3]. While early methods for creating strand-specific libraries were laborious and inefficient, technical advances have made this approach more accessible.
Experimental Design and Batch Effects
Proper experimental design is crucial for generating meaningful RNA-seq data. A well-designed experiment minimizes batch effects—technical variations that can occur during sample processing, RNA isolation, library preparation, or sequencing runs [10]. Strategies to mitigate batch effects include processing controls and experimental conditions together, performing RNA isolation on the same day, and sequencing compared groups in the same run [10].
Targeted RNA Sequencing
While whole transcriptome approaches are common, targeted RNA sequencing provides a cost-effective alternative that focuses on specific transcripts of interest [5]. This approach uses hybridization capture or amplicon-based methods to enrich for genes of interest, bypassing the need for rRNA depletion and enabling more streamlined data analysis [5]. Targeted approaches are particularly valuable for analyzing limited or degraded RNA samples, such as those from formalin-fixed paraffin-embedded (FFPE) tissue [5].
Applications of Bulk RNA-Seq in Research and Drug Development
Bulk RNA-seq has become an indispensable tool in biological research and pharmaceutical development with several key applications:
Differential Gene Expression Analysis: By comparing bulk gene expression profiles between different experimental conditions, researchers can identify genes that are upregulated or downregulated in disease states, following drug treatments, or across developmental stages [8].
Biomarker Discovery: RNA-seq facilitates the discovery of RNA-based biomarkers and molecular signatures for diagnosis, prognosis, or patient stratification in various diseases [8].
Pathway and Network Analysis: Investigating how sets of genes (pathways and networks) change collectively under various biological conditions provides systems-level insights into drug mechanisms and disease processes [8].
Novel Transcript Discovery: Bulk RNA-seq data can be used to annotate isoforms, non-coding RNAs, alternative splicing events, and gene fusions that may represent novel therapeutic targets [8].
Table 2: Common RNA Sequencing Methods and Their Research Applications
| RNA Sequencing Method | Description and Benefits | Common Research Applications |
|---|---|---|
| Total RNA/Whole Transcriptome | Examines coding and noncoding RNA simultaneously; suitable for novel discovery [5] | Comprehensive transcriptome annotation, novel gene discovery |
| mRNA Sequencing | Poly(A) selection to sequence all messenger RNA; identifies novel and known content [5] | Differential gene expression analysis, isoform characterization |
| Targeted RNA Sequencing | Sequences specific transcripts of interest to focus efforts and lower costs [5] | Biomarker validation, clinical assay development, large cohort studies |
| smRNA Sequencing | Isolation of small RNA to focus on noncoding RNA like microRNA [5] | miRNA profiling, regulatory network analysis |
Successful bulk RNA-seq experiments require careful selection of reagents and resources throughout the workflow:
Table 3: Essential Research Reagents and Resources for Bulk RNA-Seq
| Item | Function | Examples/Considerations |
|---|---|---|
| rRNA Depletion Kits | Remove abundant ribosomal RNA to focus sequencing on transcripts of interest [5] | Probe-based methods using biotinylated DNA or LNA probes; enzymatic approaches using RNase H [5] |
| Poly(A) Selection Kits | Enrich for messenger RNA through binding to polyadenylated tails [10] | Magnetic bead-based purification systems [10] |
| Library Preparation Kits | Convert RNA to sequencing-ready cDNA libraries with appropriate adapters [10] | Kits compatible with stranded protocols; those supporting low input amounts [10] |
| Strandedness Reagents | Preserve information about the transcriptional orientation of reads [3] | Chemical modification methods; direct RNA ligation approaches [3] |
| Quality Control Instruments | Assess RNA integrity and library quality before sequencing [10] | Instruments measuring RNA Integrity Number (RIN); capillary electrophoresis for library size distribution [10] |
| Reference Transcriptomes | Provide standardized gene annotations for read alignment and quantification | Ensembl, GENCODE, or species-specific databases |
| Bioinformatics Pipelines | Process raw sequencing data into interpretable results [6] [9] | Nextflow workflows like nf-core/rnaseq; command-line tools like STAR, Salmon; R packages like DESeq2, limma [6] [9] |
The evolution from Sanger sequencing to next-generation sequencing has fundamentally transformed transcriptomic research, with bulk RNA-seq emerging as a powerful, accessible technology for comprehensive gene expression analysis. While the core principles of RNA sequencing remain consistent—converting RNA to cDNA and determining its sequence—the massively parallel nature of NGS has enabled unprecedented scale, sensitivity, and discovery power. For research and drug development professionals, understanding both the technical foundations and practical considerations of bulk RNA-seq is essential for designing robust experiments, interpreting complex datasets, and advancing our understanding of biological systems and disease mechanisms. As sequencing technologies continue to evolve toward even higher throughput and longer read lengths, bulk RNA-seq will maintain its critical role in bridging population-level gene expression patterns with functional insights into cellular processes.
Bulk RNA sequencing (RNA-Seq) is a powerful next-generation sequencing (NGS) technique designed to measure the presence and abundance of ribonucleic acid molecules within a biological sample. This method provides a comprehensive transcriptome-wide profile by analyzing pooled RNA extracted from tissues or large collections of cells, offering an average gene expression snapshot across thousands to millions of cells [11] [12]. The fundamental power of bulk RNA-Seq lies in its ability to capture a broad dynamic range of expression, enabling the sensitive and accurate measurement of gene expression from both known and novel genomic features without the need for predesigned probes [12]. This technique has revolutionized transcriptomics by allowing researchers to investigate transcriptional activity, identify differentially expressed genes between conditions, and discover new RNA variants, thereby providing critical insights into gene regulation, disease mechanisms, and cellular responses to various stimuli [6] [11].
Unlike single-cell RNA-Seq which profiles individual cells, bulk RNA-Seq analyzes the collective RNA from a population, making it an indispensable tool for understanding overall transcriptomic changes in tissues, blood samples, or sorted cell populations. Its applications span from basic biological discovery to clinical drug development, where it is utilized for target identification, drug effect assessment, biomarker discovery, and mode-of-action studies [13]. The technology is particularly valuable in drug discovery workflows, where it helps researchers understand expression patterns in response to treatment, dose-response to compounds, and drug combination effects [13]. By providing both qualitative and quantitative data across the entire transcriptome, bulk RNA-Seq has become a cornerstone of modern genomic research, enabling scientists to detect transcript isoforms, gene fusions, single nucleotide variants, and other features that were previously challenging to identify [12].
Core Technical Principles
From Sample to Sequencing Library
The journey of bulk RNA-Seq begins with sample collection and preparation, where RNA is extracted from biological sources such as tissues, blood, or pooled cells. The quality and integrity of the input RNA are critical factors that significantly impact downstream results. For standard mRNA sequencing, the average library insert size should exceed 200 base pairs, with specific recommendations for read length (minimum 50 base pairs) and sequencing depth (typically 20-30 million aligned reads per replicate for robust statistical power) [11] [14].
Library preparation follows RNA extraction and involves several key steps to convert RNA into a sequence-ready format. The two primary approaches include:
- mRNA enrichment: Utilizing oligo dT beads to selectively capture polyadenylated transcripts from total RNA [12]
- rRNA depletion: Removing abundant ribosomal RNAs from total RNA samples to enable sequencing of both polyadenylated and non-polyadenylated RNAs [12]
The choice between these methods depends on research objectives. mRNA enrichment is suitable for studying protein-coding genes, while rRNA depletion provides a more comprehensive view of the transcriptome, including non-coding RNAs. For large-scale studies, particularly in drug discovery using cell lines, extraction-free RNA-Seq library preparation directly from lysates can save time and resources while handling larger sample numbers efficiently [13].
Strandedness is another crucial consideration in library preparation. Stranded RNA-Seq protocols preserve information about which DNA strand (sense or antisense) generated a transcript, enabling researchers to distinguish transcripts from overlapping genes, identify antisense sequences, and improve novel transcript annotation [12]. Experimental controls, including artificial spike-in RNAs such as SIRVs or ERCC mixes, are often incorporated to monitor technical performance, quantify RNA levels between samples, assess dynamic range, and serve as quality control metrics, especially in large-scale experiments [13] [11].
Sequencing and Alignment Fundamentals
Following library preparation, samples are sequenced using NGS platforms, generating millions of short DNA reads that correspond to fragments of the original RNA molecules. The resulting FASTQ files contain both the sequence data and associated quality scores, representing the raw data input for bioinformatic processing [6] [11].
The bioinformatic workflow addresses two primary levels of uncertainty in RNA-Seq analysis: determining the transcript of origin for each read, and converting these assignments into accurate count data [6]. Two principal computational approaches have emerged to address these challenges:
Alignment-based methods: Tools like STAR (Splice Aware Aligner) perform formal alignment of sequencing reads to either a reference genome or transcriptome, recording exact coordinates of sequence matches and mismatches [6] [11]. This approach generates SAM/BAM format files that detail alignment locations and scores, providing valuable data for extended quality checks but requiring significant computational resources.
Pseudoalignment methods: Tools such as Salmon and kallisto use lightweight algorithms that employ substring matching to probabilistically determine a read's origin without base-level alignment precision [6]. This approach is substantially faster than traditional alignment and simultaneously addresses both levels of uncertainty—read origin assignment and count estimation—while maintaining high accuracy.
A hybrid approach has gained popularity in best-practice workflows, combining the strengths of both methods. This involves using STAR for initial alignment to generate comprehensive quality control metrics, followed by Salmon operating in alignment-based mode to leverage its sophisticated statistical models for handling uncertainty in converting read origins to counts [6]. This combination provides both robust quality assessment and accurate quantification.
Table 1: Key Computational Tools for Bulk RNA-Seq Analysis
| Analysis Step | Tool Options | Primary Function | Key Considerations |
|---|---|---|---|
| Read Trimming & QC | fastp, Trim Galore, Trimmomatic | Remove adapter sequences, low-quality bases | Fastp offers speed; Trim Galore integrates FastQC for quality reports [15] |
| Alignment | STAR, HISAT2, TopHat | Map reads to reference genome/transcriptome | STAR is splice-aware; preferred for junction mapping [6] [11] |
| Quantification | Salmon, kallisto, RSEM, HTSeq | Estimate transcript/gene abundance | Salmon uses probabilistic modeling; fast and accurate [6] |
| Differential Expression | DESeq2, limma | Identify statistically significant expression changes | Choice affects false discovery rates; consider data characteristics [6] [14] |
Standardized Processing Workflow
End-to-End Analysis Pipeline
A standardized bulk RNA-Seq processing workflow transforms raw sequencing data into biologically interpretable results through a series of interconnected steps. Modern best practices often utilize automated pipelines such as the nf-core RNA-seq workflow or the ENCODE Uniform Processing Pipeline, which ensure reproducibility and consistency across analyses [6] [11].
The workflow begins with quality control and read trimming, where adapter sequences and low-quality bases are removed using tools like fastp or Trim Galore. This critical first step improves subsequent mapping rates by eliminating technical artifacts that could interfere with alignment [15]. The quality of both raw and processed data should be assessed using metrics such as Q20/Q30 scores (representing base call accuracy of 99% and 99.9% respectively) and GC content distribution [15].
Following quality control, reads undergo alignment to a reference genome using splice-aware aligners like STAR, which account for intron-exon boundaries by employing specialized algorithms to detect splicing events. For organisms without high-quality reference genomes, alignment can be performed against transcriptome sequences instead. The output of this step is a BAM file containing genomic coordinates for each successfully mapped read [6] [11].
The next critical phase is quantification, where reads are assigned to genomic features (genes, transcripts, or exons) based on annotation files (GTF/GFF format). This process generates the fundamental data structure for downstream analysis: a count matrix with rows representing features and columns representing samples [6] [11]. Tools like RSEM (RNA-Seq by Expectation Maximization) employ statistical models to account for reads that map ambiguously to multiple genes or isoforms, while Salmon uses a lightweight-alignment approach to estimate transcript abundances [6].
The final analytical stage involves differential expression analysis using specialized statistical methods implemented in tools like limma or DESeq2. These approaches model count data using appropriate statistical distributions (typically negative binomial) to identify genes exhibiting significant expression differences between experimental conditions while controlling for multiple testing [6] [14]. The output is a list of differentially expressed genes (DEGs) with associated statistical measures (p-values, false discovery rates, and fold changes), which serve as the basis for biological interpretation.
Quality Assurance and Normalization
Robust quality assurance is essential throughout the RNA-Seq pipeline to ensure reliable results. The ENCODE consortium has established comprehensive standards for bulk RNA-Seq experiments, including requirements for replicate concordance (Spearman correlation >0.9 between isogenic replicates), minimum read depths (30 million aligned reads per replicate), and metadata completeness [11].
Multiple quality metrics should be examined, including:
- Alignment rates: The percentage of reads that successfully map to the reference
- Genomic distribution: The proportion of reads mapping to exonic, intronic, and intergenic regions
- 3' bias: Potential preferential coverage at the 3' end of transcripts, particularly relevant for degraded samples
- Duplicate reads: Artifactual amplification of specific fragments
- RRNA contamination: Excessive mapping to ribosomal RNA genes
Following quantification, normalization is critical to remove technical variations and enable meaningful comparisons between samples. Different normalization strategies address distinct aspects of technical bias:
- Library size normalization: Accounts for differences in total sequencing depth between samples
- Gene length normalization: Corrects for the fact that longer genes naturally accumulate more reads (implemented in FPKM/RPKM/TPM)
- Compositional normalization: Addresses the fact that a small number of highly expressed genes can consume substantial sequencing depth
The most commonly used normalized units include:
- CPM: Counts Per Million - simple library size normalization
- FPKM: Fragments Per Kilobase of transcript per Million mapped reads - adjusts for both library size and gene length
- TPM: Transcripts Per Million - a more robust version of FPKM that accounts for the distribution of transcript lengths
Table 2: Standard Quantitative Outputs from Bulk RNA-Seq Analysis
| Output Metric | Calculation | Application | Considerations |
|---|---|---|---|
| Raw Counts | Number of reads mapping to a feature | Primary input for differential expression analysis | Most statistically rigorous for DE testing [6] |
| CPM | Counts per million mapped reads | Basic cross-sample comparison | Does not account for gene length differences [16] |
| FPKM/RPKM | Fragments per kilobase per million | Gene expression normalization | Not comparable across samples [11] |
| TPM | Transcripts per million | Most reliable normalized unit | Comparable across samples [11] |
| Expected Counts | Probabilistic estimates accounting for multi-mapping | Input for differential expression | Generated by tools like Salmon, RSEM [6] |
Experimental Design Considerations
Replication and Statistical Power
Proper experimental design is paramount for generating biologically meaningful RNA-Seq data. A fundamental consideration is statistical power—the probability of detecting genuine differential expression when it truly exists. Underpowered experiments with insufficient replicates remain a prevalent issue in transcriptomics, with approximately 50% of human RNA-Seq studies using six or fewer replicates per condition, and 90% of non-human studies falling at or below this threshold [14].
The relationship between replicate number and statistical power is complex, influenced by effect sizes (magnitude of expression differences), biological variability, and sequencing depth. Empirical evidence suggests that a minimum of six biological replicates per condition is necessary for robust detection of differentially expressed genes, increasing to twelve or more replicates when comprehensive DEG detection is required [14]. While financial and practical constraints often limit replication numbers, researchers should prioritize biological replicates over technical replicates, as the former capture natural variation between individuals, tissues, or cell populations, while the latter primarily assess technical variation from sequencing runs or laboratory workflows [13] [14].
Pilot studies are highly valuable for determining appropriate sample sizes for main experiments by providing preliminary data on variability. Consulting with bioinformaticians during the planning phase can help researchers optimize the trade-off between cohort size, sequencing depth, and budget constraints [13]. For studies involving precious or limited samples, such as patient biopsies, researchers should employ statistical methods specifically designed for small sample sizes and interpret results with appropriate caution regarding false discovery rates [14].
Batch Effects and Confounding Factors
Batch effects—systematic technical variations introduced by processing samples at different times, locations, or personnel—represent a significant challenge in RNA-Seq studies. These non-biological variations can confound results if not properly addressed in the experimental design [13]. Large-scale studies inevitably incur batch effects as samples cannot be processed simultaneously due to logistical constraints.
Several strategies can mitigate batch effects:
- Randomization: Distributing samples from different experimental conditions across processing batches
- Blocking: Intentionally grouping similar samples together in processing batches
- Balanced design: Ensuring each batch contains proportional representation of all experimental conditions
- Batch correction algorithms: Statistical methods that remove technical variations during data analysis
Plate layout should be carefully planned to facilitate later batch correction in silico if complete randomization is impossible [13]. Additionally, spike-in controls provide an internal standard for normalizing between batches and monitoring technical performance across large experiments [13] [11].
Other important design considerations include:
- Time points: For time-course experiments, multiple time points should be included to capture dynamic gene expression changes
- Controls: Appropriate negative and positive controls should be incorporated, particularly in drug treatment studies
- Sample quality: RNA integrity number (RIN) should be assessed, with minimum thresholds established prior to sequencing
- Blinding: Whenever possible, sample processing and data analysis should be performed blinded to experimental conditions to minimize bias
Applications in Research and Drug Discovery
Bulk RNA-Seq has become an indispensable tool across biological research and pharmaceutical development due to its comprehensive transcriptome-wide profiling capabilities. In basic research, applications include characterizing transcriptional landscapes, identifying novel genes and splice variants, studying gene regulation, and understanding developmental processes [15] [12].
In the drug discovery and development pipeline, RNA-Seq is applied at multiple stages [13]:
- Target identification: Discovering novel disease-associated genes and pathways
- Mechanism of action studies: Elucidating how drug candidates affect cellular processes
- Biomarker discovery: Identifying expression signatures predictive of drug response
- Toxicology studies: Understanding off-target effects and compound toxicity
- Clinical trial stratification: Defining patient subgroups based on molecular profiles
The integration of bulk RNA-Seq with emerging technologies is expanding its applications further. For example, combining bulk transcriptomics with single-cell RNA-Seq enables researchers to contextualize findings at cellular resolution, distinguishing whether expression changes occur uniformly across cell types or are specific to particular subpopulations [16] [17]. Computational deconvolution methods leverage single-cell RNA-Seq references to estimate cellular proportions from bulk data, extending the utility of existing bulk RNA-Seq datasets [16] [17].
As a genomic resource, bulk RNA-Seq continues to contribute to large-scale mapping projects such as the Genotype-Tissue Expression (GTEx) project and the Human Protein Atlas, which provide reference expression patterns across normal human tissues [17]. These resources enable researchers to interpret disease-associated genes in the context of normal tissue expression, identify tissue-specific drug targets, and understand the molecular basis of tissue specificity.
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 3: Essential Materials and Reagents for Bulk RNA-Seq Experiments
| Reagent/Resource | Function | Examples & Considerations |
|---|---|---|
| RNA Extraction Kits | Isolate high-quality RNA from biological samples | Choose based on sample type (cells, tissues, blood, FFPE); assess recovery of RNA species of interest [13] |
| Library Prep Kits | Prepare RNA for sequencing | Stranded mRNA vs. total RNA kits; consider 3'-end methods (QuantSeq) for large screens [13] [12] |
| Spike-in Controls | Monitor technical performance & normalize | ERCC RNA Spike-In Mix; SIRVs; use at ~2% of final mapped reads [11] |
| rRNA Depletion Kits | Remove abundant ribosomal RNA | Critical for total RNA sequencing; enables detection of non-polyadenylated transcripts [12] |
| Reference Genomes | Sequence alignment and quantification | ENSEMBL, UCSC; requires matching GTF/GFF annotation files [6] [11] |
| Quality Control Tools | Assess RNA and library quality | Bioanalyzer, Fragment Analyzer; FastQC for sequence data [15] |
This technical guide provides a comprehensive overview of the core terminology and methodologies underlying bulk RNA sequencing (RNA-seq). Framed within a broader thesis on how bulk RNA sequencing works as a research tool, this document details the key concepts of reads, transcriptomes, and expression quantification. It is structured to equip researchers, scientists, and drug development professionals with the foundational knowledge required to design, interpret, and critically evaluate bulk RNA-seq experiments, thereby enabling robust biological discovery and translational application.
Core Terminology and Definitions
In bulk RNA-seq, the transcriptome refers to the complete set of RNA transcripts in a biological sample, representing the functional output of the genome at a given time [18]. The process involves sequencing millions of these RNA fragments, generating reads, which are the short digital sequences determined by the instrument [10]. The primary goal is expression quantification, which estimates the abundance of each gene or transcript from the collected reads [6].
Quantitative Metrics for Expression
The table below summarizes the standard metrics used for quantifying gene expression from bulk RNA-seq data.
Table 1: Standard Gene Expression Quantification Metrics
| Metric | Full Name | Calculation | Use Case |
|---|---|---|---|
| Count | Raw Count | Number of reads uniquely assigned to a gene. | Primary input for statistical tests in differential expression analysis (e.g., with DESeq2) [19] [20]. |
| FPKM | Fragments Per Kilobase of transcript per Million mapped reads | Count of fragments (for paired-end) or reads (for single-end) per kilobase of transcript length per million mapped reads. | Normalizes for gene length and sequencing depth; allows for comparison across different genes within a sample. |
| TPM | Transcripts Per Million | Proportional count of transcripts per million transcripts in the sample. | Normalizes for gene length and sequencing depth; considered more robust than FPKM for cross-sample comparison [18]. |
| CPM | Counts Per Million | Raw counts scaled by the total number of reads per sample (in millions). | Simple normalization for sequencing depth; does not account for gene length differences. |
The Bulk RNA-Seq Workflow: From Sample to Data
The bulk RNA-seq process is a multi-step protocol that converts biological samples into interpretable gene expression data.
Experimental Protocol
A generalized, detailed methodology is as follows:
- Sample Preparation and RNA Isolation: Cells or tissues are homogenized, and total RNA is isolated using methods like column-based kits or TRIzol reagents, with care taken to prevent RNA degradation [18]. For specific cell types, such as human neutrophils, isolation via negative selection kits is recommended to minimize activation [21]. RNA quality is assessed using instruments like a Bioanalyzer (generating an RNA Integrity Number, RIN >7.0 is often required) or Nanodrop [10] [18].
- Library Preparation: This step prepares the RNA for sequencing. While protocols vary, a common workflow includes:
- mRNA Enrichment: Poly(A)+ RNA is selected using magnetic beads, or ribosomal RNA (rRNA) is depleted [10] [22].
- cDNA Synthesis: RNA is reverse-transcribed into more stable complementary DNA (cDNA). In some protocols, early barcoding is used, where sample-specific DNA tags are incorporated during this step, allowing for later pooling of samples [23].
- Fragmentation and Adapter Ligation: The cDNA is fragmented (unless using a fragmentation-free protocol), and sequencing adapters are ligated to the ends. These adapters are essential for binding to the sequencing flow cell and for sample identification [10] [18] [22].
- PCR Amplification: The final library is amplified by PCR to introduce indices and generate sufficient material for sequencing [22].
- Sequencing: The pooled libraries are loaded onto a high-throughput platform (e.g., Illumina), which generates millions of short reads (typically 50-300 base pairs) per sample [18]. The output is raw data in the form of FASTQ files, which contain the nucleotide sequences and associated quality scores for each read [18] [6].
Computational Analysis Protocol
Once FASTQ files are generated, a bioinformatics pipeline is employed:
- Quality Control (QC): Raw reads in FASTQ files are assessed for quality, adapter contamination, and correct nucleotide distribution using tools like FastQC. Trimming tools like Trimmomatic or Cutadapt are used to remove low-quality bases and adapters [19] [18].
- Read Mapping/Alignment: The cleaned reads are aligned to a reference genome or transcriptome using splice-aware aligners like STAR or HISAT2. This step identifies the genomic origin of each RNA fragment [10] [11] [18].
- Expression Quantification: Reads that are uniquely mapped to genes are counted using tools like HTSeq-count or featureCounts, generating a count matrix where rows are genes and columns are samples [10] [19] [18]. Alternatively, alignment-free tools like Salmon use pseudoalignment to rapidly quantify transcript abundance [6] [20].
- Differential Expression Analysis: The count matrix is analyzed with statistical tools like DESeq2 or edgeR to identify genes that are significantly differentially expressed between experimental conditions (e.g., treated vs. control) [10] [19] [18]. These tools internally normalize counts to account for differences in library size and apply statistical models (e.g., negative binomial distribution) to test for significance [19].
Diagram 1: Bulk RNA-seq workflow from sample to analysis.
Visualizing Expression Quantification Concepts
The process of going from sequenced reads to a quantified transcriptome involves several conceptual steps that account for technical biases and biological variation.
Diagram 2: Core steps in expression quantification and normalization.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of a bulk RNA-seq experiment relies on a suite of specialized reagents, tools, and software.
Table 2: Essential Research Reagents and Tools for Bulk RNA-seq
| Category | Item | Function and Description |
|---|---|---|
| Wet-Lab Reagents | Poly(A) Selection or Ribo-depletion Kits | Enriches for messenger RNA (mRNA) by targeting poly(A) tails or removing abundant ribosomal RNA (rRNA) [10] [22]. |
| Reverse Transcriptase Enzyme | Synthesizes complementary DNA (cDNA) from the RNA template, a critical first step in library preparation [10] [23]. | |
| Library Preparation Kit (e.g., NEBNext, TruSeq) | Provides optimized enzymes and buffers for the end-repair, adapter ligation, and amplification steps to create sequencing-ready libraries [10] [23]. | |
| DNase I | Digests and removes contaminating genomic DNA from the RNA sample to ensure only RNA is sequenced [21] [23]. | |
| ERCC Spike-in Controls | Synthetic RNA molecules added at known concentrations to the sample, used as an external standard for evaluating technical sensitivity and accuracy of quantification [11]. | |
| Bioinformatics Tools | Quality Control Tools (FastQC, Trimmomatic) | Assess read quality and perform trimming to remove low-quality bases and adapter sequences [19] [18]. |
| Aligner (STAR, HISAT2) | Precisely maps sequencing reads to a reference genome, accounting for spliced transcripts [10] [11] [18]. | |
| Quantification Tool (HTSeq-count, featureCounts, Salmon) | Counts the number of reads mapped to each gene or transcript, generating the raw count matrix [10] [19] [6]. | |
| Differential Expression Tool (DESeq2, edgeR, limma) | Performs statistical analysis on the count matrix to identify significantly differentially expressed genes between conditions [10] [19] [6]. |
Bulk RNA sequencing (bulk RNA-Seq) is a powerful technique for measuring the average gene expression levels in a sample comprising a pooled population of cells or an entire tissue section [24] [25]. Its utility spans comparative transcriptomics, biomarker discovery, and understanding physiological and pathological mechanisms [26] [25]. The fundamental value of a bulk RNA-Seq experiment is not determined by the sequencing technology itself, but by the robustness of its experimental design. A carefully crafted design, with clearly defined objectives and meticulously planned sample groups, is the cornerstone for generating meaningful, reproducible, and biologically interpretable data. This is especially critical in applied fields like drug discovery, where RNA-Seq is used at various stages, from target identification to studying drug effects and treatment responses [13]. This guide outlines the core principles of defining objectives and sample groups within the broader context of how bulk RNA sequencing works, providing a framework for researchers to build successful experiments.
Defining the Research Objective and Hypothesis
The initial and most crucial step in any bulk RNA-Seq experiment is to establish a clear research objective and a testable hypothesis. This foundational work guides every subsequent decision, from the choice of model system to the depth of sequencing and the statistical methods for analysis [13].
Formulating the Core Question
A well-defined objective should specify the biological system, the conditions being compared, and the expected outcome. The hypothesis provides a specific, testable statement about gene expression changes under these conditions. A clear aim is essential to guide the experimental design, from the chosen model system and experimental conditions to the library preparation method and sequencing setup [13].
- Example Objective: To identify differentially expressed genes in liver tissue between a wild-type mouse model and a mouse model with a specific gene knockout.
- Example Hypothesis: The knockout of Gene X will lead to the significant upregulation of genes involved in the inflammatory response and downregulation of genes in the fatty acid metabolism pathway.
Aligning Objectives with Bulk RNA-Seq Capabilities
Bulk RNA-Seq is ideally suited for hypotheses concerning the average transcriptional profile of a cell population or tissue. Researchers should consider if their project requires a global, unbiased readout or if a targeted approach is more suitable [13]. Key questions to consider include:
- Is the biological question focused on the overall state of the tissue, or is it concerned with cellular heterogeneity?
- What type of RNA data is needed? Is the focus on quantitative gene expression, or are qualitative aspects like isoform usage, splice variants, or novel transcripts also of interest? [13]
- Does the experimental design offer the flexibility for future data mining? [13]
For investigations where cellular heterogeneity is a key factor, bulk RNA-Seq may be complemented or replaced by single-cell approaches. However, bulk RNA-Seq remains highly valuable, especially when studying homogenous cell populations, when sample suspension for single-cell analysis is difficult, or when the budget necessitates a larger number of replicates and conditions [23].
Fundamentals of Sample Group Design
Once the objective is defined, the next step is to design the sample groups that will robustly test the hypothesis. This involves defining conditions, controls, and determining the sample size with adequate replication.
Conditions and Controls
A typical bulk RNA-Seq experiment compares two or more conditions. The key is to design these groups to minimize confounding factors—where the effects of two different sources of variation cannot be distinguished [27].
- Treatment vs. Control: The most common design compares a treatment group (e.g., drug-treated cells) to an untreated control group. It is critical that the control group is appropriate and that animals or samples in each condition are matched for sex, age, litter, and batch wherever possible [27] [13].
- Avoiding Confounding: An experiment is confounded if you cannot separate the effects of your variable of interest from another, unaccounted-for variable. For example, if all control mice are female and all treatment mice are male, the effect of the treatment is confounded by sex [27]. To avoid this, ensure subjects are equally split between conditions for all known variables like sex, age, and batch [27].
- Experimental Controls: In addition to biological controls, artificial spike-in RNA controls can be valuable tools. These are added in known quantities to each sample and serve as an internal standard to measure the performance of the assay, including its dynamic range, sensitivity, and reproducibility [13].
The Critical Role of Replication
Replication is non-negotiable for a statistically sound bulk RNA-Seq experiment. It is essential for accounting for natural variation and ensuring findings are reliable and generalizable [27] [13].
Table 1: Types of Replicates in Bulk RNA-Seq
| Replicate Type | Definition | Purpose | Example |
|---|---|---|---|
| Biological Replicate | Independent biological samples for the same condition (e.g., different individuals, animals, or cell cultures) [13]. | To measure biological variability and ensure findings are reliable and generalizable [13]. | Liver tissue from three different mice in each experimental group (treatment vs. control) [13]. |
| Technical Replicate | The same biological sample measured multiple times through the experimental workflow [13]. | To assess and minimize technical variation from sequencing runs, lab workflows, or the environment [13]. | Taking the same RNA sample and preparing three separate sequencing libraries from it [13]. |
For differential expression analysis, biological replicates are absolutely essential [27]. While technical replicates were more common with older technologies like microarrays, the technical variation in modern RNA-Seq is much lower than biological variation, making technical replicates largely unnecessary [27]. The primary focus should be on maximizing the number of independent biological replicates.
Sample Size and Statistical Power
The number of biological replicates per group (sample size) directly impacts the statistical power of the experiment—the ability to detect genuine differential expression. While large sample sizes are ideal, they are often constrained by cost and sample availability [13].
- General Guidelines: For general gene-level differential expression, at least 3 biological replicates per condition are typically recommended [27] [13]. However, between 4-8 replicates per group are ideal for covering most experimental requirements and for increasing reliability when variability is high [13].
- Replicates vs. Sequencing Depth: Increasing the number of biological replicates generally provides more statistical power than increasing the sequencing depth per sample. The following diagram illustrates this relationship, showing that more replicates tend to return more differentially expressed genes than deeper sequencing [27].
Figure 1: The relative importance of biological replicates versus sequencing depth for identifying differentially expressed genes. Adapted from Liu, Y., et al., Bioinformatics (2014) [27].
- Consulting Specialists: It is always beneficial to consult a bioinformatician or data expert to discuss the study and sample size limitations concerning statistical power [13]. Pilot studies are an excellent way to assess preliminary data on variability and help determine the appropriate sample size for the main experiment [13].
Advanced Considerations: Batch Effects and Confounding
Even with well-defined groups and replicates, hidden technical artifacts can undermine an experiment. The most significant of these are batch effects.
Understanding and Managing Batch Effects
Batch effects are systematic, non-biological variations introduced by how samples are processed [13]. They can arise from differences in RNA isolation date, library preparation date, personnel, reagent lots, or equipment [27]. The effect of batches on gene expression can often be larger than the experimental effect of interest [27].
- Identifying Batches: If the answer to any of the following questions is 'No', you have batches [27]:
- Were all RNA isolations performed on the same day?
- Were all library preparations performed on the same day?
- Did the same person perform the RNA isolation/library prep for all samples?
- Did you use the same reagents for all samples?
Best Practices for Batch Management
- Avoid Confounding by Batch: The most critical rule is to never confound your experiment by batch. Do NOT process all samples from one condition in one batch and all samples from another condition in a separate batch [27].
- Balance Across Batches: Split replicates of the different sample groups across batches [27]. For example, if you have three treatment groups (A, B, C) and can only process six samples per day, you should process at least one sample from each group in every batch.
- Record Metadata: Always include comprehensive batch information in your experimental metadata. During the statistical analysis, this information can be used to regress out the variation due to batch, provided the design is not confounded [27].
Technical Specifications and the Scientist's Toolkit
With the biological design in place, attention must turn to the technical specifications that will support the research objectives.
Sequencing Depth and Read Length
The optimal sequencing depth and read length depend on the specific aims of the study. The following table summarizes general guidelines.
Table 2: Technical Specifications for Different Bulk RNA-Seq Applications
| Application | Recommended Sequencing Depth | Recommended Read Length | Key Considerations |
|---|---|---|---|
| General Gene-level DE | 15-30 million single-end reads per sample [27]. | >= 50 bp [27]. | 15 million reads may be sufficient with >3 replicates; ENCODE suggests 30M [27]. |
| DE of Lowly Expressed Genes | 30-60 million reads per sample [27]. | >= 50 bp [27]. | Start with 30 million reads if you have a good number of replicates [27]. |
| Isoform-level DE (Known isoforms) | At least 30 million reads per sample; paired-end reads required [27]. | >= 50 bp; longer is better [27]. | Choose biological replicates over deeper sequencing [27]. |
| Isoform-level DE (Novel isoforms) | > 60 million reads per sample [27]. | Longer reads are beneficial (e.g., from PacBio or Oxford Nanopore) [26]. | Provides improved coverage for identifying new splicing events [26]. |
Research Reagent Solutions and Essential Materials
A successful bulk RNA-Seq experiment relies on a suite of specialized reagents and materials. The following table details key items and their functions.
Table 3: Essential Research Reagents and Materials for Bulk RNA-Seq
| Item | Function | Examples / Notes |
|---|---|---|
| RNA Extraction Kit | Isolates total RNA from cells or tissue. | Must be suitable for sample type (e.g., cell lines, blood, FFPE). Some kits retain small RNAs [13]. |
| DNase I | Digests genomic DNA contaminants during RNA purification. | Prevents amplification of genomic DNA, which is a particular concern for protocols capturing intronic reads [23]. |
| Poly(dT) Oligos / Beads | Enriches for polyadenylated mRNA from total RNA by binding the poly-A tail. | Used when the focus is on mature, protein-coding mRNA [26] [12]. |
| Ribo-depletion Reagents | Selectively depletes ribosomal RNA (rRNA) from total RNA. | Used for total RNA-seq, allowing quantification of non-polyadenylated RNAs and pre-mRNA [26] [7]. |
| Spike-in RNA Controls | Artificial RNA sequences added in known quantities to each sample. | Serves as an internal standard for normalization, quality control, and assessing technical performance [13]. |
| Reverse Transcriptase | Synthesizes complementary DNA (cDNA) from the RNA template. | MMLV-derived enzymes are common; they can also prime DNA, highlighting the need for effective DNase treatment [23]. |
| Library Prep Kit | Prepares the cDNA for sequencing by adding platform-specific adapters. | Kits like Illumina TruSeq are standard. Early barcoding protocols (e.g., Prime-seq) can drastically reduce costs [23]. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide tags added to each molecule during cDNA synthesis. | Allows bioinformatic identification and removal of PCR duplicates, improving quantification accuracy [23]. |
The overall workflow, from sample to data, integrates these components into a coherent pipeline, as shown below.
Figure 2: A generalized bulk RNA-Seq workflow, highlighting the critical RNA quality control checkpoint [26].
Bulk RNA-Seq Workflow: From Sample to Biological Insight
In bulk RNA sequencing (RNA-Seq), the quality of the final data is profoundly determined at the very first steps: sample preparation and RNA extraction. This technical guide details the critical protocols and considerations for securing high-quality RNA, framing them within the broader context of how bulk RNA-Seq transforms biological starting material into actionable gene expression data. Bulk RNA-Seq measures the average expression level of individual genes across hundreds to millions of input cells, providing a global overview of the transcriptome from pooled cell populations, tissue sections, or biopsies [24] [12]. The integrity of this data hinges on the initial handling of source material, as minute introductions of contaminants, RNA degradation, or unintended biases during sample preparation can irrevocably compromise downstream analyses, leading to inaccurate biological interpretations. Adherence to rigorous, standardized protocols from the start is therefore not merely a preliminary step but a core determinant of the entire project's success.
Critical Pre-Extraction Considerations
The journey to quality RNA begins long before the extraction kit is opened. Several pre-analytical factors must be carefully controlled to preserve the native transcriptome.
Source Material and Handling
Source material can range from cultured cells and tissue biopsies to blood samples. A key consideration is that bulk RNA-Seq analyzes RNA pooled from a population of cells, providing a population-average expression profile [24]. Immediately upon collection, cellular RNA must be stabilized to prevent rapid degradation by ubiquitous RNases. For tissues, flash-freezing in liquid nitrogen is a standard method. Alternatively, immersion in commercial RNA stabilization reagents is highly effective, especially for longer storage or shipping [28]. Storage of stabilized samples should be at -80°C for long-term preservation [28]. It is critical to minimize the time between sample collection and stabilization, as delays can induce stress-related changes in gene expression that confound experimental results.
Experimental Design to Mitigate Batch Effects
A well-designed experiment accounts for and minimizes batch effects—technical sources of variation that are unrelated to the biological question. These effects can arise from multiple users, different days of RNA isolation, or separate sequencing runs. To mitigate this, researchers should process control and experimental samples simultaneously whenever possible, from RNA isolation through library preparation and sequencing [10]. Table 1 outlines common sources of batch effect and strategies to minimize them.
Table 1: Common Sources of Batch Effect and Mitigation Strategies
| Source Category | Specific Examples | Mitigation Strategies |
|---|---|---|
| Experimental | Multiple users; time of day; animal cage effects | Standardize protocols; harvest at same time of day; use littermate controls. |
| RNA Isolation & Library Prep | Different isolation days; technician variation; freeze-thaw cycles | Isolate RNA for all samples on the same day; minimize users. |
| Sequencing | Different sequencing lanes or runs | Sequence samples from all experimental groups on the same flow cell. |
RNA Extraction Methodologies
Selecting the appropriate RNA extraction method is crucial and depends on the sample type, required RNA species, and downstream applications.
Core Principles and Quantity Requirements
The fundamental goal of RNA extraction is to isolate total RNA that is pure, intact, and free of contaminants like genomic DNA, proteins, and salts. For standard bulk RNA-Seq library preparation, the recommended starting amount is typically between 100 ng to 1 µg of purified total RNA, with many core facilities recommending at least 500 ng [28]. Input requirements can be lower for more specialized, low-input protocols. The extracted RNA must be stored in an RNase-free environment, and its quality must be rigorously assessed before proceeding.
Comparison of Extraction Techniques
Several methods are available for RNA extraction, each with advantages and limitations. The choice of kit and method should be guided by the sample type and research needs. Table 2 provides a comparative overview of different RNA extraction technologies.
Table 2: Comparison of RNA Extraction Methods and Kits
| Best For | Product Name | Starting Material | RNA Types Isolated | Isolation Method/Format | Prep Time |
|---|---|---|---|---|---|
| Simple, reliable, rapid method | PureLink RNA Mini Kit | Bacteria, blood, cells, liquid samples | Large RNA (mRNA, rRNA) | Silica spin column | 20 min |
| Micro RNA and total RNA | mirVana miRNA Isolation Kit | Cells, tissue | Small & large RNA (microRNA, tRNA, mRNA, rRNA) | Organic extraction + spin column | 30 min |
| High-throughput applications | MagMAX for Microarrays Total RNA Isolation Kit | Blood, cells, tissue | Small & large RNA (microRNA, tRNA, mRNA, rRNA) | Plate-based + magnetic beads | <1 hr |
| mRNA sequencing | Dynabeads mRNA DIRECT Kit | Cell lysate | mRNA only | Magnetic bead capture | 15 min |
| FFPE tissue | MagMAX FFPE DNA/RNA Ultra Kit | FFPE curls | total RNA, microRNA, gDNA | Magnetic beads | 48 min (for 96 preps) |
The Scientist's Toolkit: Essential Reagents and Materials
Successful RNA extraction and library preparation rely on a suite of specialized reagents. The following table details key solutions used in the featured protocols.
Table 3: Research Reagent Solutions for RNA-Seq Sample Preparation
| Reagent / Material | Function / Explanation |
|---|---|
| RNA Stabilization Reagents (e.g., TRIzol) | Protects RNA from degradation immediately after sample collection by inactivating RNases. |
| DNase I | Enzyme that degrades residual genomic DNA during extraction to prevent DNA contamination in RNA-seq libraries. |
| Silica Spin Columns / Magnetic Beads | Solid-phase matrices that bind RNA specifically under certain buffer conditions, allowing for purification from contaminants. |
| Oligo(dT) Magnetic Beads | Used to selectively isolate polyadenylated mRNA from total RNA by binding to the poly-A tail. |
| ERCC RNA Spike-In Mixes | Synthetic RNA controls added to samples before library prep to monitor technical performance and quantify expression. |
| RiboMinus Probes | Used to selectively deplete ribosomal RNA (rRNA), which can constitute >80% of total RNA, to enrich for transcriptomic reads. |
| Unique Dual Index Adapters | DNA barcodes ligated to cDNA during library prep, allowing multiple samples to be pooled ("multiplexed") and sequenced together. |
Quality Control and Integrity Assessment
After extraction, RNA quality must be quantitatively assessed before proceeding to library construction. Two primary metrics are used:
- Concentration and Purity: Quantification using fluorometric methods (e.g., Qubit) is preferred for accuracy. Spectrophotometric measurements (A260/A280 and A260/A230 ratios) assess purity, with ideal ratios being ~2.0 for both, indicating minimal contamination from proteins or solvents [24].
- RNA Integrity: This is critical for sequencing success. The RNA Integrity Number (RIN) or equivalent metric is determined using systems like the Agilent TapeStation or Bioanalyzer. For bulk RNA-Seq, an RIN value >7.0 is generally considered the minimum threshold for high-quality data, with higher values (e.g., >8.0) being preferable [10]. Degraded RNA leads to 3' bias in sequencing libraries and reduces the power to detect full-length transcripts.
From RNA to Sequencing Library: Core Workflows
Once high-quality RNA is obtained, it is converted into a sequenceable library. The following diagram illustrates the two primary workflows for this process.
RNA Enrichment Strategies
As shown in the workflow, a key decision is the RNA enrichment strategy, which directly impacts the transcriptional features captured:
- Poly(A) Selection: This method uses oligo(dT) beads or magnetic particles to selectively isolate messenger RNA (mRNA) molecules that possess a poly-adenylated tail [12] [10]. This is the most common approach for standard gene expression profiling but will miss non-polyadenylated RNAs (e.g., some non-coding RNAs).
- rRNA Depletion: As an alternative, this method uses probes to remove abundant ribosomal RNA (rRNA), which can constitute over 80% of total RNA [28]. This retains both polyadenylated and non-polyadenylated transcripts, providing a broader view of the transcriptome, and is particularly useful for studying non-model organisms or bacterial RNA.
Library Construction and Barcoding
Following enrichment, the RNA is converted to cDNA via reverse transcription. Adapters, which include sample-specific indices (barcodes), are then ligated to the fragments [28]. These barcodes enable the pooling of dozens of samples into a single sequencing run, as the sequencer reads can later be bioinformatically sorted back to their sample of origin. Early barcoding methods, where samples are given unique identifiers during the cDNA synthesis step, have been developed to significantly improve cost-efficiency by allowing early pooling of samples [23]. The final library is amplified by PCR to generate sufficient material for sequencing.
Sample preparation and RNA extraction form the foundational pillar of any robust bulk RNA-Seq study. The meticulous attention to detail during sample stabilization, the strategic selection of an extraction methodology suited to the biological question, and the rigorous application of quality control metrics are non-negotiable prerequisites. By ensuring the integrity of the RNA from the very start, researchers lay the groundwork for generating high-fidelity gene expression data, thereby maximizing the potential for meaningful biological discovery and the advancement of therapeutic development.
RNA sequencing (RNA-seq) has instigated a transformative shift in molecular biology, enabling researchers to explore gene expression profiles and regulatory mechanisms within cells with unparalleled precision [29]. At the core of every bulk RNA-seq experiment lies the critical process of library preparation—a procedure that transcribes RNA molecules into a collection of DNA fragments appropriately structured for high-throughput sequencing platforms [29]. This technical guide examines the fundamental principles, methodologies, and practical considerations for converting RNA into sequence-ready libraries, framed within the broader context of bulk RNA sequencing workflow. Library preparation serves the multifaceted purpose of preserving biological information contained within RNA molecules while simultaneously incorporating essential adaptors and barcodes required for sequencing [29]. The quality of this initial step profoundly influences all subsequent data generation and interpretation, making its optimization essential for robust transcriptomic analysis.
Core Principles of Library Construction
The conversion of RNA to a sequence-ready library follows a defined series of molecular steps. The process initiates with RNA isolation from biological specimens, followed by fragmentation into smaller segments conducive to sequencing [29]. These RNA fragments undergo reverse transcription into complementary DNA (cDNA) using reverse transcriptase enzymes [29] [10]. The resulting cDNA fragments are processed through end repair to create blunt ends, facilitating the ligation of adaptors containing necessary sequencing motifs [30] [29]. Finally, the constructed library undergoes enrichment and size selection to isolate fragments within the desired size range, followed by rigorous quality control assessments prior to sequencing [29].
A critical innovation in modern library preparation is early barcoding, where sample-specific DNA tags are integrated during cDNA generation [23]. This approach allows pooling of samples from multiple experimental conditions early in the workflow, significantly reducing processing time and reagent costs while maintaining sample identity throughout the sequencing process.
Key Methodological Variations
Library preparation strategies diverge based on RNA transcript targets and enrichment methods:
Poly(A) Enrichment: This approach selectively captures messenger RNA (mRNA) molecules containing poly-A tails, focusing on the protein-coding transcriptome [30] [10]. The Illumina Stranded mRNA Prep kit exemplifies this method, providing cost-effective, scalable RNA sequencing of the coding transcriptome with precise strand orientation [30].
Ribosomal RNA (rRNA) Depletion: For comprehensive transcriptome analysis including non-coding RNAs, this method removes abundant ribosomal RNA through enzymatic or probe-based depletion [30]. The Illumina Stranded Total RNA Prep employs integrated enzymatic depletion to remove both rRNA and globin mRNA in a single, rapid step [30].
Targeted Enrichment: Focusing on specific genes or transcripts of interest, this approach uses hybridization-based capture to enrich particular regions prior to sequencing [30]. The Illumina RNA Prep with Enrichment enables deep insights into focused gene sets without requiring mechanical shearing [30].
Table 1: Comparison of RNA Library Preparation Methods
| Method | Primary Target | Key Applications | Input Requirements | Hands-on Time |
|---|---|---|---|---|
| mRNA Sequencing | Poly-A-containing transcripts | Gene expression quantification, isoform identification | 25-1000 ng standard quality RNA [30] | < 3 hours [30] |
| Total RNA Sequencing | Whole transcriptome (coding and noncoding) | Novel feature detection, comprehensive transcriptome analysis | 1-1000 ng standard quality RNA; 10 ng for FFPE [30] | < 3 hours [30] |
| Targeted RNA Sequencing | Specific genes/transcripts of interest | Gene fusion detection, variant identification | 10 ng standard quality RNA [30] | < 2 hours [30] |
| Prime-seq | 3' tagged transcriptome | Cost-effective gene expression profiling | Not specified | Not specified |
Detailed Experimental Protocol
RNA Isolation and Quality Control
The library preparation process begins with RNA extraction, a step requiring meticulous attention to prevent degradation:
- Sample Collection: Employ aseptic techniques for procuring tissue or cell samples, promptly transferring them into RNase-free vessels to prevent RNA degradation [29].
- Homogenization: Utilize appropriate mechanical methods (tissue homogenization or bead milling) to disrupt cellular structures and facilitate RNA liberation [29].
- RNA Extraction: Execute RNA extraction using commercial isolation kits, adhering strictly to manufacturer guidelines. Include robust DNase treatment to eliminate genomic DNA contamination [29].
- Quality Assessment: Quantify RNA concentration and purity using spectrophotometry (NanoDrop) or fluorometry (Qubit). Evaluate RNA integrity through capillary electrophoresis (Bioanalyzer) or agarose gel electrophoresis [29] [10]. High-quality RNA typically demonstrates an RNA integrity number (RIN) >7.0 for reliable library construction [10].
Library Construction Workflow
The following diagram illustrates the core workflow for converting quality-controlled RNA into sequence-ready libraries:
Diagram 1: RNA Library Preparation Workflow. This flowchart illustrates the key steps in converting isolated RNA into sequence-ready libraries, from fragmentation to final quality control.
RNA Fragmentation: Prepare fragmentation buffer according to manufacturer specifications, tailoring conditions to achieve desired fragment sizes (typically 200-500bp) [29]. Introduce isolated RNA into fragmentation buffer, incubating under specified temperature and time parameters. Terminate the reaction using stop solution or thermal inactivation [29].
cDNA Synthesis: Assemble a master mix containing reverse transcriptase enzyme, random primers, dNTPs, and RNase inhibitor. Incubate fragmented RNA within the master mix at appropriate temperatures to facilitate cDNA synthesis [29]. Purify synthesized cDNA using purification kits or magnetic beads to remove residual primers, enzymes, and salts [29].
End Repair and Adaptor Ligation: Execute end repair by treating purified cDNA with end repair enzymes and buffers to create blunt-ended fragments [29]. Prepare a ligation mix containing adaptors with unique barcodes or indices. Ligate adaptors to repaired cDNA termini under specified conditions, then purify ligated products to eliminate unligated adaptors [30] [29].
Size Selection and Amplification: Employ gel electrophoresis, bead-based purification, or automated liquid handling systems to isolate DNA fragments within the desired size range (typically 200-500bp for Illumina platforms) [29]. Amplify size-selected library fragments via PCR using primers complementary to adaptor sequences, optimizing conditions to minimize amplification bias and ensure uniform coverage [29].
Quality Control and Quantification
Rigorous quality assessment ensures library integrity before sequencing:
- Library Quantification: Employ qPCR with adaptor-specific primers or fluorometric quantification to accurately measure amplified library concentration [30] [29].
- Size Distribution Analysis: Evaluate library size distribution and integrity through capillary electrophoresis (Bioanalyzer) or agarose gel electrophoresis [29].
- Quality Metrics: Verify that libraries meet platform-specific specifications for concentration, size distribution, and adapter content. The ENCODE consortium recommends specific standards for bulk RNA-seq, including Spearman correlation >0.9 between isogenic replicates [11].
Technological Innovations in Library Prep
Tagmentation-Based Methods
A significant advancement in library preparation technology is bead-linked transposome tagmentation [30]. This innovative approach simultaneously fragments DNA and adds sequencing adapters using an engineered transposase enzyme complex, dramatically reducing hands-on time and processing steps. Tagmentation-based methods like those employed in the Illumina RNA Prep with Enrichment kit enable completion of the entire RNA or DNA workflow in a single shift, with approximately two hours of hands-on time [30].
Early Barcoding Strategies
Prime-seq exemplifies the efficient adaptation of single-cell RNA-seq principles to bulk sequencing, incorporating early barcoding to significantly reduce costs [23]. This method utilizes poly(A) priming, template switching, and unique molecular identifiers (UMIs) to generate 3' tagged RNA-seq libraries [23]. Research demonstrates that Prime-seq performs equivalently to standard methods like TruSeq but is fourfold more cost-efficient due to almost 50-fold cheaper library costs [23].
Table 2: Performance Comparison of Library Prep Methods
| Method | Cost Efficiency | Hands-on Time | Complexity | Key Advantages |
|---|---|---|---|---|
| Traditional TruSeq | Standard | 6.5-7 hours [30] | Moderate | Comprehensive coverage, established protocol |
| Tagmentation-Based | High | < 2 hours [30] | Low | Rapid protocol, minimal hands-on time |
| Prime-seq | Very High (4× TruSeq) [23] | Not specified | Moderate | Extreme cost efficiency, early barcoding |
| Stranded Total RNA | Moderate | < 3 hours [30] | Moderate | Whole transcriptome coverage, rRNA depletion |
Unique Molecular Identifiers
Unique Molecular Identifiers (UMIs) represent another critical innovation, providing error correction and enhancing accuracy by reducing false-positive variant calls while increasing variant detection sensitivity [30]. These random DNA tags incorporated during library preparation enable precise identification of PCR duplicates, essential for accurate transcript quantification, particularly in low-input scenarios [23].
The Scientist's Toolkit: Essential Research Reagents
Successful library construction requires specific reagents and materials, each serving distinct functions in the workflow:
Table 3: Essential Reagents for RNA-seq Library Preparation
| Reagent/Category | Function | Examples/Notes |
|---|---|---|
| RNA Isolation Kits | Purify RNA from biological samples | PicoPure RNA isolation kit [10], commercial kits with DNase treatment [29] |
| Poly(A) Selection Beads | Enrich for messenger RNA | NEBNext Poly(A) mRNA magnetic isolation kits [10], oligo-dT beads |
| rRNA Depletion Kits | Remove ribosomal RNA | Illumina Stranded Total RNA Prep with enzymatic depletion [30] |
| Reverse Transcriptase | Synthesizes cDNA from RNA templates | MMLV-derived enzymes, template-switching variants [23] |
| Fragmentation Reagents | Break RNA into appropriately sized fragments | Enzymatic cleavage, chemical fragmentation, or sonication [29] |
| Library Prep Kits | Comprehensive reagents for end prep and adapter ligation | NEBNext Ultra DNA Library Prep Kit [10], Illumina Stranded mRNA Prep [30] |
| Unique Dual Indexes | Enable sample multiplexing | Up to 384 UDIs for higher throughput sequencing [30] |
| Size Selection Beads | Isolate fragments within optimal size range | SPRI beads, AMPure XP beads [29] |
| Quality Control Instruments | Assess RNA and library quality | Bioanalyzer, TapeStation, Qubit fluorometer [29] [10] |
Troubleshooting and Optimization
Addressing Common Challenges
Several factors require careful consideration during library preparation optimization:
RNA Input Amount: Inadequate RNA input may cause biased library construction and reduced sequencing depth, while excess input can yield inefficient adaptor ligation and elevated background noise [29]. While 0.1-1 μg of total RNA is generally recommended, specialized kits can handle much lower inputs [29].
Fragmentation Method: The choice between sonication, enzymatic cleavage, or chemical fragmentation influences size distribution and cDNA fragment integrity [29]. Optimization should align with experimental requirements and sequencing platform specifications.
Adaptor Design: Adaptor architecture affects library complexity, sequencing coverage, and read quality [29]. Custom adaptor sequences with distinctive barcodes facilitate sample multiplexing and precise sample identification during data analysis.
Managing Technical Artifacts
Technical artifacts pose significant challenges in library preparation:
Genomic DNA Contamination: Despite DNase treatment during RNA isolation, residual genomic DNA can contribute to intronic reads [23]. Prime-seq validation experiments demonstrate that DNase I treatment effectively minimizes this contamination, confirming that most intronic reads derive from pre-mRNA rather than genomic DNA [23].
Batch Effects: Variations during experiment execution, RNA isolation, library preparation, or sequencing runs can introduce confounding batch effects [10]. Mitigation strategies include processing controls and experimental conditions simultaneously, minimizing users, and harvesting samples at consistent times [10].
RNA-seq library preparation represents the foundational step in bulk transcriptome analysis, transforming biological RNA samples into sequence-ready formats compatible with high-throughput platforms. Method selection should be guided by experimental objectives, sample characteristics, and resource constraints. Traditional poly(A)-enriched libraries remain ideal for focused mRNA analysis, while ribosomal RNA-depleted libraries enable comprehensive transcriptome characterization. Technological innovations like tagmentation and early barcoding have dramatically improved efficiency and reduced costs. Regardless of the specific method employed, rigorous quality control throughout the library preparation process remains essential for generating robust, reproducible sequencing data that powers accurate biological insights in both basic research and drug development applications.
Next-generation sequencing (NGS) has revolutionized biological research by enabling the comprehensive analysis of genetic material at an unprecedented scale. For researchers investigating transcriptomes, bulk RNA sequencing (bulk RNA-Seq) serves as a powerful technique that measures gene expression in a sample, providing large-scale insights into cellular processes by averaging signals across many cells [7]. This methodology involves converting RNA molecules into complementary DNA (cDNA) and sequencing them using sophisticated platforms [7]. The resulting data empowers scientists to compare gene expression between different conditions, discover novel transcripts, identify biomarkers, and perform pathway analyses critical for drug development and basic research [7].
High-throughput sequencing systems, particularly Illumina's NovaSeq series, have become cornerstones of modern genomics facilities due to their scalable output and robust performance. These platforms leverage proven sequencing by synthesis (SBS) technology and patterned flow cell technology to generate billions of sequencing reads in a single run [31]. For research professionals designing transcriptomic studies, understanding the capabilities, specifications, and operational considerations of these systems is paramount for generating high-quality, reproducible data that can yield meaningful biological insights within project constraints and timelines.
Comparative Analysis of High-Throughput Sequencing Platforms
Illumina NovaSeq Series: Technical Specifications
The Illumina sequencing platform portfolio offers two primary high-throughput systems: the established NovaSeq 6000 and the more recent NovaSeq X Series. These systems provide scalable solutions for large-scale transcriptomic studies.
Table 1: Comparison of Illumina High-Throughput Sequencing Platforms
| Specification | NovaSeq 6000 | NovaSeq X | NovaSeq X Plus |
|---|---|---|---|
| Maximum Output | 6 Tb (dual flow cell) | 8 Tb | 16 Tb |
| Maximum Reads per Run | 20B single reads / 40B paired-end | 26B single reads / 52B paired-end | 52B single reads / 104B paired-end |
| Maximum Read Length | 2 × 250 bp | 2 × 150 bp | 2 × 150 bp |
| Run Time | 13–44 hours | 13–48 hours | 13–48 hours |
| Quality Scores (Q30) | ≥75% to ≥90% (depends on read length) | ≥85% to ≥90% (depends on read length) | ≥85% to ≥90% (depends on read length) |
| Integrated DRAGEN Analysis | On-premises or cloud | Onboard | Onboard |
| Key Innovation | Patterned flow cell technology | XLEAP-SBS chemistry | XLEAP-SBS chemistry with higher throughput |
The NovaSeq 6000 System, with its tunable output of up to 6 Tb and support for various flow cell types (SP, S1, S2, S4), offers remarkable flexibility for different project scales [31] [33]. Its flow cells can be run singly or in pairs, with dual flow cell runs delivering twice the output. For bulk RNA-Seq applications, the system can process approximately 32-400 transcriptomes per run depending on the flow cell type, assuming ≥50 million reads per sample [31].
The newer NovaSeq X Series incorporates XLEAP-SBS chemistry, which delivers improved reagent stability with two-fold faster incorporation times compared to previous chemistry [32]. This platform also features enhanced sustainability benefits including lyophilized reagents that arrive at room temperature and a significant reduction in packaging waste. The integrated DRAGEN secondary analysis platform enables ultra-rapid, accurate genomic data analysis directly on the instrument [32].
Table 2: NovaSeq Output Specifications for Transcriptome Sequencing
| Flow Cell Type | Reads Passing Filter | Output (2×150 bp) | Estimated Transcriptomes per Flow Cell |
|---|---|---|---|
| NovaSeq 6000 S4 | 16-20B paired-end | 2400-3000 Gb | ~400 |
| NovaSeq 6000 S2 | 6.6-8.2B paired-end | 1000-1250 Gb | ~164 |
| NovaSeq X 25B | 52B paired-end | ~8 Tb | ~520 |
| NovaSeq X 10B | 20B paired-end | ~3 Tb | ~200 |
Platform Selection Considerations for Bulk RNA-Seq
Choosing the appropriate sequencing platform depends on several factors specific to each research project. The scale of the study is a primary consideration – the number of samples and required sequencing depth per sample directly influences which instrument and flow cell type will be most cost-effective. For large cohort studies with hundreds of samples, the NovaSeq X Plus with 25B flow cells offers unparalleled throughput, while smaller projects might benefit from the flexibility of NovaSeq 6000 with S2 or S1 flow cells.
Run time represents another critical factor in platform selection. A NovaSeq 6000 2×150 bp run requires approximately 25-44 hours depending on the flow cell type, while comparable runs on the NovaSeq X Series take 23-48 hours [31] [32]. These timeframes include automated cluster generation, sequencing, post-run wash, and base calling, but exclude library preparation and secondary analysis time. Projects with tight deadlines might prioritize instruments with faster turnaround times.
Data quality requirements also guide platform selection. Both NovaSeq systems deliver high-quality data, with ≥85% of bases exceeding Q30 at 2×150 bp read length [31] [32]. The NovaSeq X Series employs two-channel SBS with blue-green optics and a custom CMOS sensor for ultra-high-resolution imaging, potentially providing more consistent quality across runs [32].
Bulk RNA Sequencing Methodology
Experimental Design and Planning
Effective bulk RNA-Seq studies begin with meticulous experimental design. Researchers must first establish a clear biological question and hypothesis, which will guide subsequent decisions about sample size, sequencing depth, and analysis strategy [34]. A well-defined experimental design ensures that the resulting data will have sufficient statistical power to detect biologically meaningful differences while accounting for potential sources of technical and biological variation.
The RNA biotype of interest represents another fundamental consideration in experimental planning. While messenger RNAs (mRNAs) encoding proteins are frequently the focus, many studies also investigate non-coding RNAs including long non-coding RNAs (lncRNAs), microRNAs (miRNAs), and circular RNAs, each requiring specialized library preparation approaches [34]. Standard mRNA-Seq workflows typically employ poly-A selection to enrich for polyadenylated transcripts, but this approach will miss non-polyadenylated RNA species and may be unsuitable for degraded samples [34].
Figure 1: Bulk RNA-Seq Experimental Workflow. Key decision points (yellow) significantly impact data quality and interpretation. [7] [34]
RNA Quality Assessment and Library Preparation
RNA quality is paramount for successful bulk RNA-Seq experiments and cannot be remedied once compromised. The RNA Integrity Number (RIN) provides a quantitative measure of RNA quality, with values greater than 7 generally indicating sufficient integrity for high-quality sequencing [34]. However, this threshold may vary depending on the biological sample source. Blood samples, for instance, often present challenges in maintaining high RNA integrity and typically require collection in RNA-stabilizing reagents like PAXgene or immediate processing followed by storage at -80°C [34].
Library preparation constitutes perhaps the most technically complex aspect of bulk RNA-Seq workflows. The process typically involves reverse transcribing fragmented RNA into cDNA, adding platform-specific sequencing adapters, and often includes PCR-based amplification [34]. A critical decision in this process is whether to use stranded or unstranded protocols. Stranded libraries preserve information about the original transcript orientation, which is crucial for identifying antisense transcription, accurately quantifying overlapping genes, and determining expression isoforms generated by alternative splicing [34]. While unstranded protocols are simpler, cheaper, and require less input RNA, stranded approaches are generally preferred for their richer transcriptional information [34].
Ribosomal RNA (rRNA) depletion represents another essential consideration in library preparation, as rRNA constitutes approximately 80% of cellular RNA [34]. Without depletion, the majority of sequencing reads would map to ribosomal sequences, dramatically increasing the cost required to obtain sufficient coverage of non-ribosomal transcripts. Depletion strategies include rRNA-targeted DNA probes conjugated to magnetic beads and RNase H-mediated degradation of rRNA-DNA hybrids [34]. Each method presents trade-offs between efficiency and reproducibility that must be considered based on experimental goals.
Sequencing Configuration and Depth
Appropriate sequencing depth is critical for detecting differentially expressed genes with statistical significance while maintaining cost efficiency. For standard bulk RNA-Seq differential expression analyses, 20-50 million reads per sample often suffices, though studies focusing on low-abundance transcripts or detecting subtle expression changes may require substantially greater depth [31] [32]. The read length configuration also impacts data utility – 2×150 bp paired-end reads currently represent the standard for Illumina platforms, providing sufficient length for accurate alignment while allowing detection of alternative splicing events.
Table 3: Essential Research Reagent Solutions for Bulk RNA-Seq
| Reagent/Category | Function | Key Considerations |
|---|---|---|
| RNA Stabilization Reagents | Preserve RNA integrity during sample collection/storage | Critical for challenging samples like blood; PAXgene is exemplary |
| Poly-A Selection Beads | Enrich for polyadenylated mRNA | Excludes non-polyadenylated RNAs; requires high RNA integrity |
| rRNA Depletion Kits | Remove ribosomal RNA | Increases sequencing efficiency; choice between bead-based vs enzymatic methods |
| Stranded cDNA Synthesis Kits | Convert RNA to sequencing-ready cDNA | Preserves strand information; utilizes dUTP/second strand degradation |
| Library Amplification Mixes | Amplify adapter-ligated cDNA | PCR conditions affect bias; requires optimization |
| Quality Control Assays | Assess RNA/library quality | Bioanalyzer/TapeStation for RIN; qPCR for library quantification |
Data Management and Analysis Pipeline
NGS Data Formats and Storage Considerations
Next-generation sequencing workflows generate diverse file formats, each serving specific purposes in the analysis pipeline. Understanding these formats is essential for efficient data management and processing.
FASTQ files represent the primary output from sequencing instruments, containing raw nucleotide sequences along with per-base quality scores [35]. These text-based files can be substantial, often ranging from gigabytes to terabytes, and are typically compressed using gzip (.fastq.gz) for storage efficiency [35]. The FASTQ format includes four lines per sequence: a header beginning with "@", the nucleotide sequence, a separator line ("+"), and quality scores encoded in ASCII characters representing Phred quality values [35].
Following alignment to a reference genome, data typically converts to SAM (Sequence Alignment/Map) or its compressed binary equivalent, BAM [35]. The SAM format provides a comprehensive, human-readable representation of alignments, while BAM offers the same information in a compressed, indexed format optimized for computational efficiency and random access to specific genomic regions [35]. The newer CRAM format provides even greater compression by storing only differences from a reference sequence, potentially reducing file sizes by 30-60% compared to BAM [36]. This makes CRAM particularly valuable for long-term data archiving and large-scale projects with substantial storage requirements.
Figure 2: Bulk RNA-Seq Data Analysis Pipeline. Key file formats (green) transition from raw sequences to analysis-ready data. [7] [35]
Bulk RNA-Seq Analysis Workflow
The bulk RNA-Seq analysis pipeline involves multiple computational steps transforming raw sequencing data into biologically interpretable results. Following sequencing, quality control assessments evaluate data using tools like FastQC to identify potential issues including adapter contamination, low-quality bases, or unusual sequence content. Problematic reads may be filtered or trimmed at this stage to improve downstream analysis quality.
Read alignment to a reference genome or transcriptome represents the next critical step, with popular tools including STAR, HISAT2, and Bowtie2 efficiently mapping millions of reads to their genomic origins. The resulting alignment files (BAM/SAM format) then undergo quantification, where reads are assigned to genomic features (genes, transcripts) and counted [7] [35]. This generates a count matrix – a tabular representation of expression values (genes as rows, samples as columns) that serves as the foundation for subsequent differential expression analysis [35].
The GeneLab consortium has developed a standardized processing pipeline for bulk RNA-Seq data that identifies differentially expressed genes through a consensus approach developed with scientific community input [7]. This workflow, wrapped into a Nextflow framework for reproducibility and scalability, processes all bulk RNA-Seq datasets hosted on the Open Science Data Repository (OSDR), with processed data products publicly available alongside each dataset [7]. Such standardized approaches enhance reproducibility and comparability across studies, particularly important in large-scale transcriptomic investigations.
Emerging Trends and Future Directions
The NGS field continues to evolve rapidly, with several emerging trends poised to influence bulk RNA-Seq approaches in the near future. Multiomic integration – combining genomic, epigenomic, and transcriptomic data from the same sample – represents a powerful approach for uncovering complex biological mechanisms [37]. In 2025, population-scale genome studies are expected to expand to this new phase of multiomic analysis enabled by direct interrogation of molecules, moving beyond cDNA proxies to direct RNA and epigenome analysis [37].
Artificial intelligence and machine learning are increasingly integrated into NGS data analysis, helping researchers unravel complex biological patterns from high-dimensional datasets [37]. AI-powered analytics can accelerate biomarker discovery, refine diagnostic processes, and guide the development of targeted therapies by identifying patterns that might escape conventional analytical approaches [37]. The intersection of NGS and AI will be critical for generating the large datasets required to drive biomedical breakthroughs at scale.
Spatial transcriptomics represents another frontier, with 2025 expected to be a breakthrough year for sequencing-based technologies that enable direct sequencing of cells within their native spatial context in tissue [37]. This approach empowers researchers to explore complex cellular interactions and disease mechanisms with unprecedented biological precision, particularly when applied to clinically characterized FFPE samples [37]. While currently distinct from bulk RNA-Seq, technological advances may eventually blur the boundaries between these approaches.
The continuing decentralization of sequencing also marks an important trend, with clinical sequencing applications moving beyond central hubs to individual institutions [37]. This distribution brings sequencing closer to domain expertise, potentially accelerating insights and application of genomic medicine. Platforms like the NovaSeq X Series with their integrated analysis capabilities and simplified workflows support this trend by reducing the technical barriers to high-quality sequencing.
For researchers planning bulk RNA-Seq studies, these evolving landscapes highlight the importance of designing experiments with future integration in mind – considering how today's transcriptomic data might complement tomorrow's multiomic datasets and analytical approaches.
Bulk RNA sequencing (RNA-seq) is a foundational transcriptomic method that measures the average gene expression across a population of cells within a sample [38]. This technique provides critical insights throughout therapeutic development, enabling researchers to dissect disease mechanisms, validate drug targets, and assess therapeutic efficacy and safety from initial discovery through clinical applications [38]. The bioinformatics pipeline that transforms raw sequencing data into biologically meaningful information forms the analytical backbone of these investigations. This technical guide details the core components of this pipeline—quality control, read alignment, and expression quantification—framed within the context of how bulk RNA sequencing powers research discoveries.
Quality Control of Raw Sequencing Data
Initial Quality Assessment
The initial quality control (QC) phase is crucial for ensuring the integrity and accuracy of all downstream analyses by systematically identifying and removing poor-quality sequences and technical artifacts from raw sequencing data [38]. This process begins with generating comprehensive quality reports for the input reads using tools such as FastQC and MultiQC [39] [38].
Key quality metrics assessed include:
- Sequence quality scores across all bases
- GC content and its distribution
- Adapter contamination levels
- Overrepresented k-mers
- Duplicated read rates (potential PCR artifacts)
Typically, read quality decreases toward the 3' end of reads, and bases with low quality scores must be trimmed to improve mappability [40]. This initial QC stage generates critical baseline data that informs subsequent filtering and trimming parameters.
Read Filtering and Trimming
Following initial quality assessment, reads undergo filtering and trimming processes to remove technical sequences and low-quality bases. Commonly used tools for this stage include fastp, Trim Galore (which integrates Cutadapt and FastQC), Trimmomatic, and Cutadapt itself [15] [38].
The specific filtering and trimming operations include:
- 5' and 3' trimming of bases with low quality scores [38]
- Adapter removal to eliminate sequences originating from library preparation adapters [38]
- Read length optimization for alignment, particularly important for specific RNA types like miRNAs [38]
- Optional alignment and filtering of reads against specific genomic loci (e.g., rRNAs, tRNAs, snoRNAs) [38]
Table 1: Bioinformatics Tools for Quality Control and Trimming
| Tool | Primary Application | Key Features | Considerations |
|---|---|---|---|
| FastQC | Quality control of raw reads | Generates comprehensive quality reports; works with any sequencing platform [38] [40] | Does not perform filtering itself |
| fastp | Filtering and trimming | Rapid processing; simple operation; significantly enhances processed data quality [15] | |
| Trim Galore | Filtering and trimming | Integrates Cutadapt and FastQC; generates QC reports during processing [15] | May cause unbalanced base distribution in tail regions [15] |
| Trimmomatic | Filtering and trimming | Highly cited QC software [15] | Complex parameter setup; no speed advantage [15] |
| Cutadapt | Adapter removal | Specialized in removing adapter sequences [38] | Often integrated within other tools like Trim Galore |
Only reads that pass all filters in the data preparation stage are kept for subsequent analysis. The pipeline produces comprehensive quality reports after this phase, including FastQC reports and information about the fraction of reads aligned to various genomic loci if this option was selected [38].
Read Alignment Strategies
Alignment Approaches
Read alignment involves mapping the filtered sequencing reads to reference sequences, and there are three primary strategies for this process, each with distinct advantages and considerations [40]:
- Genome Mapping: Aligns reads to a reference genome using splice-aware aligners
- Transcriptome Mapping: Aligns reads directly to a set of transcript sequences
- De Novo Assembly: Assembles transcripts without a reference genome, used when reference sequences are unavailable
Table 2: Comparison of Read Alignment Strategies
| Strategy | Method | Advantages | Disadvantages | Recommended Depth |
|---|---|---|---|---|
| Genome-based | Alignment to a reference genome | Computationally efficient; eliminates contaminating reads; sensitive for low-abundance transcripts; can discover novel transcripts without annotation [40] | Requires high-quality reference genome [40] | ~10x coverage [40] |
| De novo Assembly | Assembly without a reference genome | No reference genome required; correct alignment to known splice sites not required; can assemble trans-spliced transcripts [40] | Computationally intensive; sensitive to sequencing errors [40] | >30x coverage [40] |
Alignment Tools and Considerations
For genome mapping, splice-aware aligners are essential to accommodate alignment gaps caused by introns. The most widely used tools include:
- STAR: Preferred for its accuracy in handling splice junctions; commonly used in standardized pipelines like the ENCODE Bulk RNA-seq pipeline [6] [11]
- HISAT2: An efficient alternative for genome mapping [38]
- TopHat2: Historically significant but largely superseded by newer tools [38]
Regardless of the alignment strategy, reads may map uniquely or be assigned to multiple positions in the reference. These "multi-mapped reads" or "multireads" present particular challenges: genomic multireads typically result from repetitive sequences or shared domains of paralogous genes, while transcriptome multi-mapping more often arises from gene isoforms [40].
The following diagram illustrates the core decision-making workflow for read alignment strategies:
Expression Quantification
Quantification Methods and Tools
Expression quantification transforms aligned reads into estimates of gene or transcript abundance. This process must account for two levels of uncertainty: identifying the most likely transcript of origin for each read, and converting read assignments to counts in a way that models the uncertainty inherent in many read assignments [6].
There are two primary approaches to quantification:
Alignment-based quantification: Uses formal alignments (BAM files) from tools like STAR and employs statistical methods to model uncertainty. Popular tools include:
Pseudoalignment: A faster approach that uses substring matching to probabilistically determine locus of origin without base-level alignment. Tools implementing this method include:
A hybrid approach that combines the advantages of both methods is often recommended. This involves using STAR to align reads to the genome to facilitate comprehensive quality control metrics, then using Salmon in alignment-based mode to perform quantification, leveraging its statistical model for handling uncertainty in converting read origins to counts [6].
Quantification Outputs
Quantification tools generate several key metrics for each gene or transcript:
- Expected counts: Raw estimates of transcript abundance [11]
- TPM (Transcripts Per Million): Within-sample normalized value that accounts for transcript length and sequencing depth [38] [11]
- FPKM (Fragments Per Kilobase of transcript per Million mapped reads): Similar to TPM but with different normalization properties [11]
For downstream differential expression analysis, the pipeline must generate a count matrix with rows corresponding to genes or transcripts and columns corresponding to samples [6]. This matrix serves as the primary input for statistical packages like DESeq2 and edgeR [41].
Integrated Workflow and The Researcher's Toolkit
End-to-End Pipeline Architecture
Comprehensive analysis pipelines integrate all processing steps into cohesive workflows. Reproducible pipeline frameworks such as Nextflow and Snakemake are commonly used to automate this multi-step process [6] [39]. Established bulk RNA-seq pipelines include:
- nf-core/rnaseq: A community-developed Nextflow workflow that incorporates best practices for quality control, alignment, and quantification [6]
- ENCODE Uniform Processing Pipeline: Standardized pipeline using STAR and RSEM for alignment and quantification [11]
- CHIRP: A state-of-the-art workflow used at the Minnesota Supercomputing Institute and featured in multiple publications [42]
- RnaXtract: A Snakemake-based pipeline that performs gene expression quantification, variant calling, and cell-type deconvolution [39]
- Via Foundry: A commercial solution that wraps quality control, alignment, quantification, and interpretation into an integrated pipeline [38]
The following workflow diagram illustrates how these components integrate in a complete bulk RNA-seq analysis pipeline:
The Researcher's Toolkit
Table 3: Essential Research Reagents and Computational Tools for Bulk RNA-seq Analysis
| Category | Tool/Resource | Function | Application Notes |
|---|---|---|---|
| Quality Control | FastQC | Quality control of raw sequencing reads | Generates comprehensive quality reports; first step in pipeline [38] [40] |
| fastp | Filtering and trimming | Rapid processing; significantly enhances data quality [15] | |
| Read Alignment | STAR | Splice-aware genome alignment | Accurate handling of splice junctions; used in ENCODE pipeline [6] [11] |
| HISAT2 | Efficient genome alignment | Alternative to STAR [38] | |
| Expression Quantification | Salmon | Transcript quantification | Fast pseudoalignment; handles assignment uncertainty [6] |
| RSEM | Alignment-based quantification | Uses expectation-maximization algorithm; models uncertainty [6] [11] | |
| kallisto | Pseudoalignment-based quantification | Efficient transcript-level quantification [39] [40] | |
| Workflow Management | nf-core/rnaseq | Automated end-to-end analysis | Reproducible Nextflow workflow; incorporates multiple tools [6] |
| Snakemake | Workflow management framework | Used by pipelines like RnaXtract for reproducible analysis [39] | |
| Reference Resources | GENCODE | Genome annotation | Provides comprehensive gene annotations for reference genomes [11] |
| ERCC Spike-Ins | Exogenous RNA controls | Creates standard baseline for RNA expression quantification [11] |
The bioinformatics pipeline for bulk RNA-seq data—encompassing quality control, read alignment, and expression quantification—forms an essential foundation for transcriptomic research. By implementing robust, standardized processing methods, researchers can transform raw sequencing data into reliable gene expression measurements that power discoveries across biological research and therapeutic development. As bulk RNA-seq continues to evolve alongside emerging technologies, its proven power, versatility, and continued impact remain undiminished, serving as a critical tool for understanding gene regulation, disease mechanisms, and therapeutic effects [38].
Bulk RNA sequencing (RNA-Seq) has revolutionized transcriptomics by enabling genome-wide quantification of RNA abundance, providing a comprehensive snapshot of the gene expression profile from a population of cells [43]. A primary objective in bulk RNA-Seq studies is the identification of differentially expressed genes (DEGs)—genes whose expression levels change significantly between different biological conditions, such as healthy versus diseased tissue or treated versus control samples [44]. Differential expression analysis forms a critical pillar in understanding the molecular mechanisms underlying phenotypic differences, facilitating discoveries in disease biomarker identification, drug development, and fundamental biological processes [43] [15].
Within this analytical landscape, DESeq2 and limma have emerged as two of the most widely used and robust statistical frameworks for DEG detection. DESeq2 employs a negative binomial modeling approach specifically designed for count-based RNA-Seq data, while limma, initially developed for microarray analysis, utilizes linear models with empirical Bayes moderation on transformed count data [45] [46]. This technical guide provides an in-depth examination of both tools, offering detailed methodologies, comparative analysis, and practical implementation protocols to empower researchers in generating biologically relevant insights from their transcriptomic data.
Foundational Concepts in Bulk RNA-Seq Analysis
From Raw Sequencing to Biological Insight
The journey from raw sequencing reads to a list of biologically relevant genes involves a multi-step computational workflow. After sequencing, the initial raw data in FASTQ format undergoes quality control to identify technical artifacts such as adapter contamination or poor-quality bases [43] [47]. Tools like FastQC or Falco generate quality reports, following which trimming tools such as Trimmomatic or fastp remove problematic sequences [44] [15]. The cleaned reads are then aligned to a reference genome or transcriptome using splice-aware aligners like STAR or HISAT2, or alternatively, pseudo-aligned using tools like Salmon or Kallisto [43] [6]. The aligned reads are subsequently quantified to generate a count matrix—a table where rows represent genes, columns represent samples, and values indicate the number of reads assigned to each gene in each sample [44] [47]. This count matrix serves as the fundamental input for differential expression analysis.
The Critical Role of Normalization
Raw count data cannot be directly compared between samples due to technical variations, primarily sequencing depth (the total number of reads per sample) and library composition (the distribution of reads across genes) [43]. Normalization procedures mathematically adjust the counts to remove these biases, enabling valid cross-sample comparisons. DESeq2 employs a median-of-ratios method, which calculates a size factor for each sample by comparing gene counts to a sample-specific reference [43]. In contrast, edgeR (often used in comparisons with these tools) typically uses the Trimmed Mean of M-values (TMM) method, which similarly corrects for composition bias [43]. Other methods like Counts Per Million (CPM) and Transcripts Per Million (TPM) offer simpler normalization but are generally not recommended for between-sample differential expression analysis due to their sensitivity to highly expressed genes [43].
Table 1: Common Normalization Methods in RNA-Seq Analysis
| Method | Sequencing Depth Correction | Library Composition Correction | Suitable for DE Analysis? | Primary Implementation |
|---|---|---|---|---|
| CPM | Yes | No | No | edgeR, limma (for transformation) |
| RPKM/FPKM | Yes | Yes | No | Various quantification tools |
| TPM | Yes | Yes | No | Various quantification tools |
| Median-of-Ratios | Yes | Yes | Yes | DESeq2 |
| TMM | Yes | Yes | Yes | edgeR |
Statistical Frameworks: DESeq2 and limma
DESeq2: Negative Binomial Modeling for Count Data
DESeq2 operates on the fundamental principle that RNA-Seq count data follows a negative binomial distribution, which appropriately models the over-dispersion (extra-Poisson variation) common in sequencing data [45] [48]. Its analytical process involves several key steps:
- Estimation of Size Factors: Normalizes for sequencing depth using the median-of-ratios method [43].
- Dispersion Estimation: For each gene, DESeq2 estimates the dispersion, which represents the variance of the count data around its mean. This is crucial for accounting for biological variability between replicates. DESeq2 uses an empirical Bayes shrinkage approach to stabilize dispersion estimates across genes, particularly beneficial for experiments with small sample sizes [45].
- Generalized Linear Model (GLM) Fitting: DESeq2 fits a negative binomial GLM to the normalized counts for each gene. The model includes the experimental design (e.g., ~ condition) to test for differential expression [48].
- Hypothesis Testing and Shrinkage: Using the fitted model, DESeq2 performs Wald tests or likelihood ratio tests to compute p-values. It also applies an adaptive shrinkage to log2 fold changes (LFC) to prevent large LFC estimates from genes with low counts and high dispersion, thereby reducing false positives and improving interpretability [45].
DESeq2's requirement for biological replicates is rooted in this statistical framework, as replicates are essential for reliably estimating gene-wise dispersion and biological variability [48].
limma-voom: Linear Modeling with Precision Weights
Initially developed for microarray data, limma (Linear Models for Microarray Data) was adapted for RNA-Seq data through the voom (variance modeling at the observational level) transformation [45] [6]. The limma-voom pipeline involves:
- Data Transformation: The raw counts are converted to log2-counts per million (log-CPM) using the
voomfunction. This transformation makes the data more amenable to linear modeling, as the distribution of log-CPM values becomes approximately normal [45]. - Precision Weighting: A key innovation of the
voomfunction is that it calculates precision (or uncertainty) weights for each individual observation (each gene in each sample). These weights account for the mean-variance relationship in the data, giving less weight to low-count genes (which have higher variance) and more weight to high-count genes (which have lower variance) in the linear model [45] [6]. - Linear Modeling and Empirical Bayes Moderation: The transformed and weighted data is then fit using standard linear models. Limma applies an empirical Bayes method to moderate the standard errors of the estimated log2-fold changes. This step borrows information across all genes to produce more stable and reliable inferences, especially powerful in experiments with a small number of replicates [45] [46].
The core distinction lies in their approach: DESeq2 directly models the raw counts with a distribution specific to RNA-Seq, while limma-voom transforms the data to fit a linear modeling framework suitable for continuous, normally distributed data, while carefully accounting for the characteristics of count data through precision weights.
Comparative Analysis and Tool Selection
Statistical and Performance Characteristics
The choice between DESeq2 and limma depends on the specific experimental context, as each tool has distinct strengths and performance characteristics.
Table 2: Comparative Analysis of DESeq2 and limma
| Aspect | DESeq2 | limma-voom |
|---|---|---|
| Core Statistical Approach | Negative binomial GLM with empirical Bayes shrinkage for dispersion and LFC | Linear modeling of log-CPM values with precision weights and empirical Bayes moderation of standard errors |
| Data Input | Raw, non-normalized counts | Raw counts (transformed internally by voom) |
| Variance Handling | Models gene-wise dispersion with shrinkage | Precision weights based on mean-variance trend |
| Ideal Sample Size | Moderate to large (≥3 replicates, performs better with more) [45] | Small to moderate (≥3 replicates) [45] |
| Best Use Cases | Experiments with high biological variability, subtle expression changes, strong FDR control [45] | Complex experimental designs (multi-factor, time-series), integration with other omics data [45] |
| Computational Efficiency | Can be intensive for large datasets [45] | Very efficient, scales well [45] |
| Key Strengths | Robust for low-count genes, automatic outlier detection, independent filtering [45] | Handles complex designs elegantly, works well with other high-throughput data [45] |
| Potential Limitations | Conservative fold change estimates, can be computationally intensive [45] | May not handle extreme overdispersion as well, requires careful QC of voom transformation [45] |
Concordance and Reproducibility
Despite their different statistical foundations, DESeq2 and limma often show a remarkable level of agreement in the DEGs they identify, especially in well-designed experiments with adequate replication. This concordance strengthens confidence in the resulting biological conclusions [45]. Furthermore, implementations like InMoose in Python have demonstrated nearly identical results to the original R packages for both limma and DESeq2, ensuring reproducibility and interoperability across programming environments [46].
Experimental Protocols and Implementation
Experimental Design Best Practices
The reliability of any differential expression analysis is fundamentally constrained by the quality of the experimental design.
- Biological Replicates: Are essential. They allow for the estimation of biological variance, which is critical for the statistical models in both DESeq2 and limma. While analysis with two replicates is technically possible, the ability to estimate variability and control false discovery rates is greatly reduced. A minimum of three replicates per condition is standard, though more may be required for heterogeneous samples or to detect subtle expression changes [43].
- Sequencing Depth: Affects the ability to detect expressed genes, especially those with low abundance. For standard differential expression analysis in mammalian systems, 20–30 million reads per sample is often sufficient [43]. The required depth should be guided by pilot experiments, existing datasets, or power analysis tools.
- Avoiding Single Replicates: A single replicate per condition does not allow for robust statistical inference of differential expression and should be avoided for hypothesis-driven experiments [43].
A Practical Protocol for DESeq2 in R
The following code provides a detailed protocol for performing differential expression analysis with DESeq2.
A Practical Protocol for limma-voom in R
The following code provides a detailed protocol for performing differential expression analysis with limma and the voom transformation.
Successful execution of a bulk RNA-Seq experiment and subsequent differential expression analysis relies on a suite of computational tools and resources.
Table 3: Essential Computational Tools for RNA-Seq Analysis
| Tool/Resource | Function | Use Case/Explanation |
|---|---|---|
| FastQC / Falco | Quality Control | Generates reports on raw read quality, base composition, adapter contamination [44] [47]. |
| Trimmomatic / fastp | Read Trimming | Removes adapter sequences and low-quality bases from raw reads [47] [15]. |
| STAR / HISAT2 | Read Alignment | Splice-aware aligners that map reads to a reference genome [43] [47]. |
| Salmon / Kallisto | Pseudo-alignment | Rapid, alignment-free quantification of transcript abundances [43] [6]. |
| FeatureCounts | Read Quantification | Generates the count matrix from aligned reads [47]. |
| DESeq2 | Differential Expression | Identifies DEGs using negative binomial models [45] [48]. |
| limma | Differential Expression | Identifies DEGs using linear models on transformed data [45] [6]. |
| R / RStudio | Computing Environment | The primary platform for statistical analysis and visualization [6] [47]. |
| Python (InMoose) | Computing Environment | A Python alternative for running limma, edgeR, and DESeq2 workflows [46]. |
| Reference Genome (FASTA) | Reference Sequence | The genomic sequence of the organism under study [6] [47]. |
| Annotation File (GTF/GFF) | Genomic Annotation | File defining the coordinates of genes, transcripts, and exons [6] [47]. |
DESeq2 and limma represent two powerful, yet distinct, statistical paradigms for identifying biologically relevant genes from bulk RNA-Seq data. DESeq2's strength lies in its dedicated negative binomial model for count data, providing robust performance across a wide range of conditions, particularly where biological variability is high. Limma-voom excels in its computational efficiency and flexibility, handling complex experimental designs with elegance. The choice between them is not a question of which is universally superior, but which is most appropriate for a given experimental context. Furthermore, their frequent concordance validates findings and bolsters confidence in the resulting biological insights. By leveraging the detailed protocols, comparative analyses, and resource toolkit provided in this guide, researchers and drug development professionals can strategically apply these tools to unravel the transcriptomic underpinnings of disease, treatment response, and fundamental biology.
Functional enrichment analysis is an essential step in the interpretation of data generated by bulk RNA sequencing (RNA-seq), a powerful technique that measures gene expression across a population of cells within a biological sample [8] [7]. The central challenge in bulk RNA-seq analysis lies in moving beyond simple lists of differentially expressed genes (DEGs) to extract meaningful biological insights. This is where functional enrichment methods become critical, as they provide a systematic framework to determine whether certain biological functions, pathways, or processes are over-represented in a set of genes identified from a differential expression analysis [49]. These approaches allow researchers to translate statistical findings into biological understanding by leveraging curated knowledge bases that categorize genes based on their known functions, interactions, and pathways.
The core value of functional enrichment analysis lies in its ability to contextualize omics data within existing biological knowledge. For bulk RNA-seq studies, which provide an average gene expression profile across all cells in a sample [8], enrichment analysis helps answer the crucial "so what?" question after identifying hundreds or thousands of DEGs. By determining whether functions related to translation, splicing, or specific disease pathways are statistically enriched, researchers can generate hypotheses about the underlying biology driving the observed expression changes [49]. However, it is vital to recognize that these tools suggest genes and pathways that may be involved with the condition of interest; they should not be used to draw definitive conclusions without experimental validation [49].
This technical guide focuses on three foundational approaches for functional enrichment analysis: Gene Ontology (GO) term enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and Gene Set Enrichment Analysis (GSEA). Each method offers distinct advantages and operates on different statistical principles, making them suitable for various research scenarios and question types.
Bulk RNA-Seq: The Foundation for Functional Insights
Bulk RNA-seq is a next-generation sequencing-based method that measures the whole transcriptome across a population of cells [8]. The experimental workflow begins with RNA extraction from a biological sample (e.g., cells, tissues, or whole organisms), followed by steps to enrich for messenger RNA (mRNA) – typically through polyA-selection – or deplete ribosomal RNA (rRNA) [7]. The purified RNA is then converted to complementary DNA (cDNA), and sequencing libraries are prepared for high-throughput sequencing. After sequencing, the raw data undergoes quality control, alignment to a reference genome, and gene quantification to generate a count matrix where each row represents a gene and each column represents a sample [19].
A critical characteristic of bulk RNA-seq is that it provides an average expression profile across all cells in the sample. While this offers a holistic view of the transcriptional state, it cannot resolve cell-to-cell heterogeneity [8]. This averaging effect has important implications for functional interpretation, as enriched pathways might reflect contributions from multiple cell types present in the sample.
From Raw Data to Biological Meaning
The typical analytical pipeline for bulk RNA-seq progresses through several stages before reaching functional enrichment analysis. Following raw data processing, differential expression analysis identifies genes that show statistically significant differences between experimental conditions (e.g., diseased vs. healthy, treated vs. control). Tools like DESeq2 are commonly used for this purpose, employing statistical tests that account for the count-based nature of RNA-seq data and multiple testing issues [19]. The output is typically a list of DEGs with associated statistics including log2 fold-changes, p-values, and adjusted p-values (q-values).
This DEG list serves as the primary input for functional enrichment analysis. The fundamental question these analyses address is: "Are there specific biological themes that occur more frequently in this gene list than we would expect by chance alone?" The following sections detail how GO, KEGG, and GSEA approaches answer this question through different statistical frameworks and biological databases.
Core Concepts: GO, KEGG, and Gene Sets
Gene Ontology (GO)
The Gene Ontology project provides a structured, controlled vocabulary for describing gene functions in a species-independent manner [49] [50]. This collaborative effort addresses the need for consistent descriptions of gene products across biological databases. The GO system organizes functional knowledge into three distinct ontologies:
- Biological Process (BP): Refers to larger biological objectives accomplished by multiple molecular activities, such as "transcription," "signal transduction," or "apoptosis." A biological process generally involves a chemical or physical change of the starting material or input [49].
- Molecular Function (MF): Represents the biochemical activities of individual gene products, such as "ligand," "GTPase," or "transporter." These activities are typically defined at the molecular level [49].
- Cellular Component (CC): Describes the locations in the cell where gene products are active, such as "nucleus," "lysosome," or "plasma membrane" [49].
Each GO term has a unique alphanumeric identifier (e.g., GO:0005125), a term name, and a definition. The ontologies are structured as hierarchical directed acyclic graphs, meaning terms can have multiple parent and child terms, with relationships ranging from general ("parent" terms) to specific ("child" terms) [49]. This structure allows for analysis at different levels of functional specificity.
KEGG Pathways
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database resource that integrates information on genomes, biological pathways, diseases, and chemical substances [50]. Unlike GO, which focuses on individual gene functions, KEGG emphasizes pathways – organized networks of molecular interactions that represent specific biological processes. KEGG pathways include metabolic pathways, signaling pathways, cellular processes, and disease pathways.
KEGG pathway enrichment analysis helps researchers place differentially expressed genes into the context of known biological systems, potentially revealing which entire pathways are affected in a given condition rather than just isolated functions [50]. This systems biology perspective is particularly valuable for understanding complex phenotypic changes.
Gene Set Collections and MSigDB
The Molecular Signatures Database (MSigDB) serves as the most comprehensive repository of gene sets for enrichment analysis [51] [52]. MSigDB organizes gene sets into several collections, with the most commonly used being:
- C5 (GO Gene Sets): Collection of GO terms for biological processes, molecular functions, and cellular components.
- C2 (Curated Gene Sets): Gene sets collected from various sources including online pathway databases (KEGG, Reactome) and published studies.
- H (Hallmark Gene Sets): Well-defined biological states or processes with coherent expression changes, particularly useful for cancer studies.
- C7 (Immunologic Signatures): Gene sets representing cell states and perturbations in immunologic biology [51].
These curated collections provide the foundation for both over-representation analysis and GSEA, enabling researchers to test their gene lists against thousands of biologically meaningful gene sets.
Methodological Approaches to Enrichment Analysis
Over-Representation Analysis (ORA)
Over-representation analysis (ORA) is the simplest and most direct approach for functional enrichment. ORA determines whether genes from a pre-defined list of significant genes (typically DEGs with p-values below a threshold) are over-represented in any pre-defined gene sets compared to what would be expected by chance [49] [50]. The statistical foundation for ORA is typically the hypergeometric test or Fisher's exact test [49].
The hypergeometric distribution models the probability of drawing a specific number of "successes" (genes in both your significant list and the gene set of interest) from a finite population without replacement. In mathematical terms, the probability of k successes follows the formula:
P(X=k) = [C(K,k) × C(N-K, n-k)] / C(N,n)
Where:
- N = total number of genes in the background (e.g., all genes in the genome)
- K = total number of genes associated with a specific functional category
- n = number of genes in the user's significant gene list
- k = number of genes that are both in the user's list and associated with the functional category [49]
This test yields a p-value for each functional category, which is then adjusted for multiple testing (e.g., using Benjamini-Hochberg False Discovery Rate) [49]. ORA implementations are available in tools like clusterProfiler and Enrichr [50] [51].
Gene Set Enrichment Analysis (GSEA)
Gene Set Enrichment Analysis (GSEA) takes a fundamentally different approach from ORA. Rather than using a predetermined significance threshold to create a gene list, GSEA uses all genes from an experiment ranked by their expression difference between two biological states [52] [51]. The method then examines whether members of a gene set tend to occur toward the top or bottom of this ranked list, indicating coordinated differential expression in that biological pathway.
The key advantages of GSEA include:
- No arbitrary cutoff: Uses information from all genes, not just those above a significance threshold
- Sensitivity to subtle effects: Can detect situations where all genes in a pathway show small but coordinated changes
- Directional information: Identifies whether a pathway is up- or down-regulated [52]
The GSEA algorithm computes an Enrichment Score (ES) that reflects the degree to which a gene set is overrepresented at the extremes (top or bottom) of the ranked list. Statistical significance is determined by comparing the observed ES to a null distribution generated by permuting the gene labels [52] [51]. The result includes a Normalized Enrichment Score (NES) and false discovery rate (FDR) for each gene set.
Competitive vs. Self-Contained Tests
A crucial conceptual distinction in enrichment analysis methodology is between competitive and self-contained tests [51]:
- Competitive Tests: Compare the genes in the test set against all other genes not in the set (the "background"). The null hypothesis is that the test set is at most as enriched as the background genes. ORA and GSEA are both competitive tests.
- Self-Contained Tests: Examine whether genes in the test set show any evidence of differential expression without reference to other genes. The null hypothesis is that no genes in the set are differentially expressed. Methods like ROAST are self-contained tests [51].
This distinction affects the interpretation of results. Competitive tests ask "is this pathway more affected than other pathways?" while self-contained tests ask "is this pathway affected at all?"
Table 1: Comparison of Functional Enrichment Methodologies
| Feature | Over-Representation Analysis (ORA) | Gene Set Enrichment Analysis (GSEA) |
|---|---|---|
| Input Requirements | List of significant genes (e.g., DEGs with p < 0.05) | Ranked list of all genes (typically by fold-change or significance) |
| Statistical Foundation | Hypergeometric test or Fisher's exact test | Kolmogorov-Smirnov-like running sum statistic |
| Key Output | Adjusted p-value for each enriched term | Normalized Enrichment Score (NES) and FDR |
| Major Advantage | Simple, intuitive, works with small gene lists | No arbitrary cutoff, detects subtle coordinated changes |
| Major Limitation | Depends on significance threshold, loses information from ranking | Requires larger sample sizes, computationally intensive |
| Primary Tools | clusterProfiler, Enrichr | GSEA software, fgsea [49] [52] [51] |
Experimental Protocols and Implementation
GO Over-Representation Analysis with clusterProfiler
The following step-by-step protocol demonstrates how to perform GO over-representation analysis using the clusterProfiler R package, a widely used tool for functional enrichment [49] [50]:
Step 1: Preparation of Input Data
- Generate differential expression results using tools like DESeq2 [19]
- Extract significant genes based on adjusted p-value threshold (e.g., padj < 0.05)
- Prepare background gene set (typically all genes tested in the experiment)
- Convert gene identifiers to Ensembl IDs for compatibility
Step 2: Running GO Enrichment Analysis
- Specify the organism database (OrgDb), ontology (BP, MF, or CC), and multiple testing correction method
Step 3: Interpretation and Visualization of Results
- Generate statistical summary and create publication-quality visualizations
This analysis produces a table of significantly enriched GO terms with statistics including gene counts, p-values, adjusted p-values, and the specific genes contributing to each term's significance.
GSEA Protocol with Ranked Gene Lists
For GSEA analysis, the protocol differs significantly due to the ranked-list approach:
Step 1: Gene Ranking
- Create a ranked list of all genes based on their association with the phenotype
- The ranking metric is typically log2 fold-change or signal-to-noise ratio
Step 2: Run GSEA Algorithm
- Use either the standalone GSEA software or R implementations like fgsea
- Specify the gene set collection (e.g., MSigDB Hallmark, KEGG, GO)
Step 3: Interpret GSEA Results
- Identify gene sets with significant NES (usually |NES| > 1.5) and FDR < 0.25
- Examine enrichment plots to verify clear patterns at the top or bottom of the ranked list
- Note the direction of enrichment (positive NES indicates up-regulation, negative indicates down-regulation)
Integrated Workflow for Comprehensive Analysis
A robust functional analysis strategy often combines multiple approaches:
- Start with ORA for a straightforward assessment of significantly enriched terms in DEGs
- Complement with GSEA to detect subtle coordinated changes across entire pathways
- Validate findings through experimental approaches and cross-reference with literature
- Use multiple gene set collections (GO, KEGG, Hallmark) to gain different biological perspectives
The following diagram illustrates the complete workflow from bulk RNA-seq to functional interpretation:
Visualization and Interpretation of Results
Visualizing GO Enrichment Results
Effective visualization is crucial for interpreting functional enrichment results. clusterProfiler offers several plotting options, each highlighting different aspects of the data:
- Dot Plot: Shows the number of genes associated with each term (dot size) and the p-adjusted values (color). This efficiently displays the top enriched terms by gene ratio [49].
- Enrichment Map: Clusters related GO terms together and visualizes relationships between terms, with color representing p-values and node size representing the number of significant genes [49].
- Gene-Concept Network: Displays the connections between genes and GO terms, showing which genes contribute to multiple terms.
For GSEA results, the characteristic enrichment plot displays the running enrichment score for the gene set versus the ranked list of genes, showing where the gene set members appear in the ranking.
Statistical Interpretation Guidelines
Proper interpretation of enrichment results requires attention to multiple statistical factors:
- Multiple Testing Correction: Always consider adjusted p-values (FDR) rather than raw p-values to control false positives [19]. The standard threshold is FDR < 0.05.
- Effect Size Measures: For ORA, the gene ratio (number of significant genes in set / total genes in set) provides context for the biological importance beyond statistical significance.
- Background Selection: The choice of background genes significantly impacts ORA results. Typically, all genes tested in the experiment should be used rather than the entire genome [49].
- Reproducibility: Look for consistent enrichment across multiple analytical approaches (ORA and GSEA) and related gene sets.
Avoiding Common Interpretation Pitfalls
Several common pitfalls can lead to misinterpretation of enrichment results:
- Overinterpreting Marginal Findings: Terms with just barely significant p-values and small gene counts may be false positives.
- Ignoring Technical Artifacts: Batch effects or sampling biases can create spurious enrichment patterns.
- Circular Reasoning: Using the same data for hypothesis generation and confirmation without independent validation.
- Overlooking Directionality: In GSEA, noting whether pathways are up- or down-regulated is crucial for biological interpretation.
Table 2: Essential Research Reagents and Computational Tools for Functional Enrichment Analysis
| Resource Type | Specific Tool/Database | Primary Function | Key Applications |
|---|---|---|---|
| Gene Set Databases | Gene Ontology (GO) | Structured vocabulary for gene function annotation | Categorizing genes by BP, MF, CC [49] [50] |
| KEGG Pathways | Database of biological pathways | Pathway-centric enrichment analysis [50] | |
| MSigDB | Comprehensive collection of gene sets | GSEA with curated biological signatures [52] [51] | |
| Analysis Software | clusterProfiler | R package for ORA and visualization | GO and KEGG enrichment analysis [49] [50] |
| GSEA Software | Standalone application for GSEA | Pre-ranked gene set enrichment analysis [52] | |
| fgsea | Fast R implementation of GSEA | Efficient enrichment analysis of ranked lists [51] | |
| Supporting Tools | DESeq2 | Differential expression analysis | Identifying DEGs from bulk RNA-seq [19] |
| org.Hs.eg.db | Organism-specific annotation database | Gene identifier conversion and annotation [49] | |
| Cytoscape | Network visualization | Visualizing complex pathway relationships [53] |
Advanced Applications and Integrative Approaches
Combining Single-Cell and Bulk RNA-Seq Data
Recent advances enable integrative analysis combining single-cell RNA-seq (scRNA-seq) with bulk RNA-seq data. This approach leverages the cell-type resolution of scRNA-seq with the statistical power of bulk sequencing. As demonstrated in gastric cancer research, scRNA-seq can identify cell-type-specific DEGs, which can then be used to build predictive models validated in bulk datasets [53]. This integration helps resolve the cellular heterogeneity that confounds bulk RNA-seq interpretation.
The fundamental difference between these technologies is important: bulk RNA-seq provides a population-average expression profile, while scRNA-seq profiles individual cells, revealing cellular heterogeneity and rare cell populations [8]. Functional enrichment can be performed on both data types, but scRNA-seq enables cell-type-specific pathway analysis.
Machine Learning Integration
Machine learning approaches are increasingly combined with functional enrichment to build predictive models and identify robust biomarkers. For example, in prostate cancer research, multiple machine learning algorithms have been applied to select genes from functional categories to construct prognostic signatures with clinical utility [54]. These integrated approaches can identify meta-programs – coordinated gene expression patterns that span multiple pathways – that drive disease progression and therapy resistance.
Temporal and Spatial Enrichment Analysis
Advanced enrichment methods can incorporate temporal dynamics through pseudotime analysis in single-cell data or time-course bulk experiments. Similarly, spatially-resolved transcriptomics enables enrichment analysis with anatomical context. These approaches move beyond static snapshots to reveal how pathway activation changes over time or across tissue regions.
The following diagram illustrates the relationship between different enrichment analysis methods and their applications:
Functional enrichment analysis through GO, KEGG, and GSEA provides an essential bridge between statistical findings from bulk RNA-seq data and meaningful biological interpretation. Each method offers complementary strengths: ORA gives straightforward identification of over-represented functions in significant gene lists, while GSEA detects more subtle, coordinated changes without arbitrary significance thresholds. The integration of these approaches, along with emerging methods that combine single-cell and bulk sequencing data, continues to enhance our ability to extract biological insights from transcriptomic studies.
As these methodologies evolve, several principles remain constant: the importance of appropriate statistical controls, the value of multiple complementary approaches, and the necessity of experimental validation. By applying these enrichment analysis techniques thoughtfully and critically, researchers can maximize the biological knowledge gained from bulk RNA-seq experiments and generate robust hypotheses for further investigation.
Bulk RNA sequencing (RNA-seq) has emerged as a foundational tool in molecular biology, enabling comprehensive profiling of gene expression patterns across tissue samples, cell populations, and whole organisms. This technical guide explores the transformative application of bulk RNA-seq in disease research, with particular emphasis on cancer biology, where it facilitates the discovery of molecular mechanisms, biomarker identification, and therapeutic target validation. By measuring the averaged gene expression across cell populations, researchers can compare diseased and healthy states to identify differentially expressed genes, unravel dysregulated pathways, and characterize tumor microenvironments. Framed within the broader thesis of how bulk RNA sequencing works in research, this review provides detailed methodologies, data analysis frameworks, and practical implementation guidelines to empower researchers and drug development professionals in leveraging this powerful technology for precision medicine applications.
Bulk RNA sequencing is a widely adopted technique that measures gene expression in samples consisting of large pools of cells, such as tissue sections or blood aliquots [6]. The methodology involves converting RNA molecules into complementary DNA (cDNA) and sequencing them using next-generation sequencing platforms, typically after removing ribosomal RNA (rRNA) which constitutes over 80% of total RNA [7]. This approach provides powerful, large-scale insights into gene expression, enabling systematic comparisons between different physiological conditions (e.g., healthy vs. diseased, treated vs. untreated) [7] [18]. Unlike single-cell approaches that profile individual cells, bulk RNA-seq generates averaged expression profiles across the entire cell population within a sample, offering a comprehensive overview of transcriptional activity that captures dominant expression patterns relevant to disease states [55].
In the context of disease research, bulk RNA-seq has become indispensable for uncovering molecular mechanisms driving pathology. The transcriptome provides a detailed snapshot of cellular activity, revealing which genes are active, how strongly they are expressed, and how these patterns change during disease progression or in response to therapeutic interventions [55]. While bulk RNA-seq cannot resolve cellular heterogeneity within tissues, its cost-effectiveness and analytical maturity make it particularly suitable for large-scale cohort studies, clinical trial biomarker analysis, and diagnostic development where population-level insights are clinically actionable [55] [56]. The technology has evolved significantly from early microarray-based methods, offering unprecedented precision, dynamic range, and capacity to discover novel transcripts and splicing variants without prior sequence knowledge [18] [55].
Technical Foundations of Bulk RNA-Seq
Sequencing Technologies and Evolution
The development of bulk RNA-seq has been propelled by continuous advances in sequencing technologies, which have dramatically enhanced our ability to study RNA with increasing accuracy, throughput, and cost-effectiveness. The evolution of these technologies has shaped contemporary transcriptomics research:
- Sanger Sequencing: Developed in the 1970s, this was the first method to read DNA and RNA sequences using chain-terminating nucleotides. While providing high accuracy, it was limited by low throughput and slow processing, making it unsuitable for transcriptome-wide studies [18].
- Next-Generation Sequencing (NGS): Revolutionized RNA-seq by enabling simultaneous sequencing of millions of RNA fragments. Platforms like Illumina provide high-throughput capabilities, generating comprehensive transcriptomic datasets in single experiments. NGS workflows involve fragmenting RNA, converting it to cDNA, and sequencing these fragments. The sensitivity of NGS allows detection of low-abundance transcripts and discovery of novel RNA species, making it the current standard for bulk RNA-seq [18].
- Third-Generation Sequencing (TGS): Technologies such as PacBio and Nanopore sequencing read long RNA fragments, which is particularly useful for identifying complex RNA structures and alternative splicing events. However, TGS currently has higher costs and lower accuracy compared to NGS, making it less common for routine bulk RNA-seq applications [18].
Table 1: Comparison of Sequencing Technologies for Bulk RNA-Seq
| Technology | Key Features | Advantages | Limitations | Common Applications in Disease Research |
|---|---|---|---|---|
| NGS (Illumina) | Short-read sequencing; High throughput | High accuracy; Cost-effective; Sensitive for low-abundance transcripts | Short read lengths complicate isoform resolution | Differential gene expression; Pathway analysis; Biomarker discovery |
| TGS (PacBio) | Long-read sequencing; Real-time monitoring | Direct RNA sequencing; Comprehensive isoform characterization | Higher error rate; Expensive; Lower throughput | Fusion gene detection; Alternative splicing analysis in cancer |
| TGS (Nanopore) | Long-read sequencing; Portable options | Real-time analysis; Longest read lengths; Direct RNA modifications | Higher error rate; Requires specific bioinformatics | Viral transcriptome studies; Isoform diversity in neurological disorders |
Key Technical Considerations for Disease Research
When implementing bulk RNA-seq for disease research, several technical considerations significantly impact data quality and biological interpretation:
- Sequencing Depth: The number of reads per sample determines the ability to detect low-abundance transcripts, which may include critical regulatory genes or rare splice variants. Deeper sequencing (typically 20-50 million reads per sample for standard differential expression analysis) provides more comprehensive transcriptome coverage, particularly important for detecting rare transcripts in heterogeneous tumor samples [18] [6].
- Library Preparation Strategy: The choice between ribo-depletion and polyA-selection for rRNA removal affects which RNA species are captured. PolyA-selection enriches for messenger RNA (mRNA) but excludes non-polyadenylated transcripts, while ribo-depletion retains both coding and non-coding RNAs, providing a broader view of the transcriptome [7].
- Strandedness: Strand-specific library preparation preserves the information about which DNA strand originated the RNA transcript, crucial for accurately quantifying overlapping genes and antisense transcription events often dysregulated in cancer [6].
- Sample Quality: RNA integrity number (RIN) is a critical quality metric, with values >7 generally recommended for reliable sequencing results. Degraded RNA from clinical specimens, particularly archived tissues, can introduce biases in transcript quantification [18].
Bulk RNA-Seq Workflow: From Sample to Insight
Experimental Design and Wet Laboratory Procedures
Robust experimental design forms the foundation for meaningful bulk RNA-seq studies in disease research. Careful planning at this stage ensures that results are both biologically relevant and statistically valid:
- Sample Size and Replication: Including proper biological replicates (typically 3-5 per condition) is essential for accounting biological variability and enabling powerful statistical detection of differentially expressed genes. Proper power analysis should guide sample size determination based on expected effect sizes and variability [18] [20].
- Control Groups: Appropriate control samples (e.g., healthy adjacent tissue, untreated controls, or isogenic control cell lines) are critical for distinguishing disease-specific expression changes from background variation [18].
- Batch Effects: Organizing sample processing and sequencing to minimize batch effects is crucial, as technical variability can confound biological signals. Randomizing samples across sequencing runs and including control samples in each batch facilitates statistical correction of batch effects during analysis [20].
The wet laboratory workflow for bulk RNA-seq involves several critical steps:
- Sample Preparation and RNA Extraction: Isolate RNA from samples (tissues, blood, or cell cultures) using methods like column-based kits or TRIzol reagents while preventing RNA degradation. Assess RNA quality using tools like Bioanalyzer or Nanodrop, aiming for high RNA integrity numbers (RIN >7) [18].
- Library Preparation: Convert high-quality RNA into sequencing-ready libraries through several steps:
- Reverse Transcription: Create complementary DNA (cDNA) from RNA using reverse transcriptase enzymes [7] [18].
- Fragmentation: Break cDNA into smaller fragments (typically 200-500bp) suitable for sequencing [18].
- Adapter Ligation: Add platform-specific sequencing adapters to fragment ends, enabling binding to the sequencing flow cell and sample multiplexing [18].
- Library Amplification: Use PCR to amplify the library, ensuring sufficient material for sequencing [18].
- Sequencing: Pool libraries and load onto NGS platforms (e.g., Illumina) for cluster generation and sequencing-by-synthesis. Paired-end sequencing (e.g., 2×150 bp) is recommended over single-end for more accurate transcript mapping and isoform resolution [6].
The following workflow diagram illustrates the complete bulk RNA-seq process from sample collection to data interpretation:
Bioinformatics Analysis Pipeline
The computational analysis of bulk RNA-seq data transforms raw sequencing reads into biological insights through a multi-step process. Each stage employs specialized tools and statistical methods to ensure robust and interpretable results:
- Quality Control: Assess raw sequencing data quality using tools like FastQC to evaluate read quality, detect adapter contamination, and identify overrepresented sequences. Follow with quality trimming using tools like Trimmomatic or Cutadapt to remove low-quality bases and adapter sequences [18].
- Read Mapping and Alignment: Map cleaned reads to a reference genome or transcriptome using splice-aware aligners such as STAR or HISAT2, which account for intron boundaries during alignment. For organisms without reference genomes, de novo assembly tools like Trinity can reconstruct transcriptomes from scratch [18] [6].
- Gene Expression Quantification: Quantify reads aligning to each gene using tools like featureCounts or HTSeq, generating a count matrix that represents expression levels across all genes and samples. Alternatively, alignment-free tools like Salmon use pseudoalignment to rapidly estimate transcript abundances while accounting for assignment uncertainty [18] [6].
- Normalization: Adjust raw counts to account for technical variations like sequencing depth and gene length using methods such as TPM (Transcripts Per Million), RPKM/FPKM, or DESeq2's median-of-ratios method, enabling valid comparisons between samples [18].
- Differential Expression Analysis: Identify genes showing statistically significant expression differences between conditions using tools like DESeq2, edgeR, or limma-voom. These tools employ statistical models that account for biological variability and count-based distributions to control false discovery rates [18] [6].
- Functional Enrichment Analysis: Interpret biological significance of differentially expressed genes through enrichment analysis using tools like DAVID or GSEA (Gene Set Enrichment Analysis) to identify overrepresented pathways, biological processes, or molecular functions [18].
Table 2: Essential Bioinformatics Tools for Bulk RNA-Seq Analysis
| Analysis Step | Tool Options | Key Features | Best Applications in Disease Research |
|---|---|---|---|
| Quality Control | FastQC, MultiQC | Comprehensive quality metrics; Batch reporting | Identifying low-quality samples; Detecting technical artifacts |
| Read Alignment | STAR, HISAT2 | Splice-aware; Fast processing; High accuracy | Cancer transcriptomes with alternative splicing; Fusion detection |
| Quantification | featureCounts, Salmon, kallisto | Gene/transcript-level counts; Handles ambiguity | Expression profiling; Isoform-level analysis in neurological diseases |
| Differential Expression | DESeq2, limma, edgeR | Robust statistical models; False discovery control | Identifying disease biomarkers; Treatment response signatures |
| Functional Analysis | clusterProfiler, GSEA | Pathway enrichment; Network visualization | Mechanism of action studies; Pathway dysregulation in disease |
Applications in Cancer Research
Unveiling Molecular Mechanisms and Biomarker Discovery
Bulk RNA-seq has revolutionized cancer research by enabling comprehensive molecular characterization of tumors across different stages and subtypes. By comparing gene expression profiles between tumor and normal tissues, researchers can identify dysregulated genes and pathways that drive oncogenesis, progression, and treatment resistance:
- Oncogenic Pathway Identification: Transcriptomic profiling can reveal activation of specific oncogenic signaling pathways (e.g., Wnt/β-catenin, PI3K/AKT/mTOR, MAPK) through coordinated expression changes in pathway components. This pathway-level understanding helps elucidate molecular mechanisms underlying tumor behavior and identifies potential therapeutic targets [18].
- Cancer Subtype Classification: Unsupervised clustering of bulk RNA-seq data has redefined tumor classification systems by identifying molecular subtypes with distinct clinical outcomes within histologically similar cancers. For example, breast cancer classification into luminal A, luminal B, HER2-enriched, and basal-like subtypes based on gene expression patterns has fundamentally improved prognostic stratification and treatment selection [18].
- Tumor Microenvironment Characterization: Deconvolution of bulk RNA-seq data using computational methods like CIBERSORT or ESTIMATE can infer relative proportions of immune cell populations within the tumor microenvironment, providing insights into immune contexture that predicts response to immunotherapy and prognosis [56].
- Biomarker Discovery: Differential expression analysis between treatment responders and non-responders, or between metastatic and non-metastatic cases, identifies gene expression signatures predictive of clinical outcomes. These molecular biomarkers can guide patient stratification for targeted therapies and inform prognosis [18] [56].
Bridging the DNA to Protein Divide in Precision Oncology
While DNA sequencing identifies genetic mutations present in tumors, bulk RNA-seq provides critical functional context by determining which mutations are actually transcribed and potentially translated into proteins. This "bridge between DNA and protein" makes RNA-seq particularly valuable for precision oncology applications:
- Validation of Mutational Significance: DNA-based assays detect mutations but cannot determine their functional impact. RNA-seq confirms whether DNA variants are expressed, helping prioritize clinically actionable mutations. Studies show that up to 18% of somatic single nucleotide variants detected by DNA sequencing are not transcribed, suggesting they may be clinically irrelevant [57].
- Therapeutic Target Prioritization: For targeted therapies that inhibit specific proteins, RNA evidence of mutant allele expression strengthens the rationale for treatment selection. This is particularly important for genes like EGFR, BRAF, and ALK, where targeted therapies are available [57].
- Fusion Gene Detection: RNA-seq excels at identifying gene fusions resulting from chromosomal rearrangements, which are often pathogenic drivers in cancers like leukemias, lymphomas, and sarcomas. Unlike DNA-based methods, RNA-seq can confirm fusion gene expression and identify breakpoints at base-pair resolution [57].
- Neoantigen Discovery for Immunotherapy: In personalized cancer vaccine development, RNA-seq verifies and prioritizes mutated peptides (neoantigens) that are actually expressed by tumors, enabling selection of targets most likely to elicit effective anti-tumor immune responses [57].
The following diagram illustrates how bulk RNA-seq integrates with multi-omics approaches in precision oncology:
Case Study: Application in Myeloproliferative Neoplasms
A recent study demonstrates the clinical utility of bulk RNA-seq in Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) [56]. Researchers analyzed peripheral blood and bone marrow samples from treatment-naïve patients using RNA sequencing to evaluate both genetic mutations and immune profiles. The findings revealed that bulk RNA-seq can simultaneously identify driver mutations (e.g., in JAK2, CALR, MPL) and characterize the immune landscape, including immune cell infiltration patterns and cytokine profiles. This comprehensive molecular profiling provides insights into distinct immune-related pathways involved in MPN pathogenesis and offers a cost-effective approach for routine clinical practice that could enhance personalized treatment strategies and improve prognostic accuracy [56].
Successful implementation of bulk RNA-seq in disease research requires carefully selected reagents, computational tools, and reference materials. The following table details essential components of the bulk RNA-seq workflow:
Table 3: Essential Research Reagents and Resources for Bulk RNA-Seq
| Category | Specific Items | Function/Purpose | Examples/Considerations |
|---|---|---|---|
| Sample Preparation | RNA stabilization reagents | Preserve RNA integrity during sample collection | RNAlater, PAXgene Blood RNA tubes |
| RNA extraction kits | Isolate high-quality total RNA | Column-based kits (Qiagen), TRIzol (for challenging samples) | |
| RNA quality assessment | Evaluate RNA integrity | Bioanalyzer, TapeStation, Nanodrop (RIN >7 recommended) | |
| Library Preparation | rRNA depletion kits | Remove ribosomal RNA | Ribo-Zero, RiboCop (maintains non-coding RNA) |
| polyA selection beads | Enrich for mRNA | Oligo(dT) beads (standard for mRNA sequencing) | |
| Library prep kits | Prepare sequencing libraries | Illumina TruSeq, NEBNext Ultra II (compatibility with input amount) | |
| cDNA synthesis kits | Reverse transcribe RNA to cDNA | Include reverse transcriptase, random hexamers/oligo(dT) primers | |
| Sequencing | Sequencing platforms | Generate sequence data | Illumina NovaSeq, NextSeq (balance of throughput and cost) |
| Sequencing reagents | Chemistry for sequencing | Platform-specific flow cells and sequencing kits | |
| Computational Tools | Quality control tools | Assess raw data quality | FastQC, MultiQC (critical for QC reporting) |
| Alignment software | Map reads to reference | STAR, HISAT2 (splice-aware for eukaryotic transcripts) | |
| Quantification tools | Generate expression matrix | featureCounts, Salmon (accuracy for differential expression) | |
| Differential expression | Identify significant changes | DESeq2, edgeR, limma (robust statistical frameworks) | |
| Functional analysis | Biological interpretation | clusterProfiler, GSEA (pathway and ontology enrichment) | |
| Reference Resources | Reference genomes | Mapping and annotation | ENSEMBL, GENCODE, UCSC (organism-specific versions) |
| Annotation databases | Functional annotation | Gene Ontology, KEGG, Reactome (current versions) | |
| Processing pipelines | Standardized analysis | nf-core/rnaseq, GeneLab workflow (reproducibility) |
Advanced Applications and Integration Approaches
Targeted RNA-Seq for Clinical Applications
While whole transcriptome sequencing provides comprehensive coverage, targeted RNA-seq approaches offer enhanced sensitivity for detecting specific mutations and biomarkers in clinical settings. Targeted panels focus sequencing power on genes of clinical interest, enabling deeper coverage and more reliable variant detection, particularly for rare alleles and low-abundance mutant clones [57]. For example, the Afirma Xpression Atlas (XA) targeted RNA-seq panel, which includes 593 genes covering 905 variants, is used clinically for thyroid cancer diagnosis and management [57]. Targeted approaches demonstrate particular utility when:
- Tumor Purity is Low: In samples with substantial stromal contamination, targeted sequencing achieves sufficient depth to detect mutant alleles that might be missed by whole transcriptome sequencing [57].
- Monitoring Minimal Residual Disease: The sensitivity of targeted panels enables detection of low-frequency transcripts indicative of residual disease after treatment [57].
- Validating DNA Variants: Targeted RNA-seq confirms expression of DNA-identified mutations, strengthening their clinical relevance for therapeutic decision-making [57].
Integration with Multi-Omics Approaches
Bulk RNA-seq data gains additional power when integrated with other molecular profiling data, creating a more comprehensive understanding of disease mechanisms:
- DNA-RNA Integration: Combining DNA sequencing with RNA-seq data distinguishes between silent mutations (present in DNA but not expressed) and functionally active mutations that impact the transcriptome. This integrated approach improves variant interpretation and prioritization of therapeutic targets [57].
- Proteogenomic Integration: Correlating RNA expression data with proteomic measurements validates whether transcriptional changes translate to protein-level alterations, identifying potential post-transcriptional regulation events [57].
- Epigenetic-Transcriptomic Integration: Overlaying chromatin accessibility or DNA methylation data with transcriptomic profiles reveals regulatory mechanisms driving expression changes in disease states [55].
Bulk RNA sequencing remains an indispensable tool in disease research, particularly for unraveling the molecular mechanisms underlying cancer pathogenesis and progression. Its ability to provide comprehensive, quantitative profiling of gene expression across entire transcriptomes enables researchers to identify dysregulated pathways, classify disease subtypes, discover biomarkers, and validate therapeutic targets. While emerging single-cell technologies offer unprecedented resolution for studying cellular heterogeneity, bulk RNA-seq continues to offer practical advantages for many research and clinical applications, including cost-effectiveness, analytical maturity, and suitability for large cohort studies.
The integration of bulk RNA-seq with other genomic data types, especially DNA sequencing, strengthens its utility in precision medicine by bridging the gap between genetic alterations and their functional consequences. As targeted RNA-seq approaches continue to evolve and computational methods for data analysis become more sophisticated, the clinical application of bulk RNA-seq is likely to expand, ultimately improving diagnostic accuracy, prognostic stratification, and treatment selection for patients with cancer and other complex diseases. For researchers and drug development professionals, mastering the technical foundations, analytical frameworks, and application strategies outlined in this guide provides a solid foundation for leveraging bulk RNA-seq to advance our understanding of disease mechanisms and develop more effective therapeutic interventions.
Bulk RNA sequencing (bulk RNA-seq) has established itself as a foundational technology in pharmaceutical research, providing powerful, large-scale insights into gene expression that drive therapeutic discovery and personalized medicine approaches. This method measures the average gene expression profile across a population of cells from samples such as tissues, blood, or entire model organisms, enabling comprehensive transcriptome analysis [8] [7]. Unlike single-cell approaches that resolve cellular heterogeneity, bulk RNA-seq delivers a population-level perspective that remains indispensable for many applications in drug development, particularly when investigating overall treatment effects, identifying biomarkers, and understanding pathway-level responses to therapeutic interventions [8].
The fundamental value of bulk RNA-seq in drug discovery lies in its ability to quantitatively capture transcriptome-wide changes in response to compound treatment, disease progression, or between different patient populations. Since its emergence in 2008, RNA-seq technologies have evolved substantially, with decreasing costs and standardized bioinformatics pipelines making them increasingly accessible for pharmaceutical applications [10]. In the context of personalized medicine, bulk RNA-seq enables the identification of molecular signatures that predict drug response, discover patient stratification biomarkers, and elucidate mechanisms of drug resistance [13]. This technical guide explores the experimental design, methodological considerations, and analytical frameworks for implementing bulk RNA-seq in drug development pipelines, with specific emphasis on generating actionable insights for accelerating therapeutic discovery.
Experimental Design for Drug Discovery Applications
Strategic Considerations for Robust Study Design
Careful experimental design is the most critical aspect of any RNA-seq experiment in drug discovery, as it directly impacts the reliability, interpretability, and translational potential of the resulting data. A clear hypothesis and well-defined objectives should guide the experimental design from the initial selection of model systems through to data analysis strategies [13]. Key considerations include determining whether the research question requires a global, unbiased transcriptomic readout or a more targeted approach, what magnitude of differential expression is expected, and whether the chosen model system is sufficiently responsive to reveal genuine drug-induced effects amid biological variability [13].
Sample size and statistical power significantly influence the quality and reliability of results in drug discovery projects. Statistical power refers to the ability to identify genuine differential gene expression in naturally variable datasets. While ideal sample sizes exist for optimal statistical analysis, practical constraints often include biological variation, study complexity, cost, and sample availability—particularly with precious patient specimens from biobanks [13]. For more accessible sample types like cell lines treated with compounds, larger sample sizes with increased replication are readily achievable. Consultation with bioinformaticians during the design phase is highly valuable for discussing study limitations and statistical power considerations [13]. Pilot studies represent an excellent strategy for determining appropriate sample sizes for main experiments by providing preliminary data on variability and enabling testing of multiple conditions before committing to large-scale studies [13].
Replicates, Controls, and Batch Effect Mitigation
Replicate strategies are fundamental to account for variability within and between experimental conditions in drug discovery studies:
Table 1: Replicate Strategies in RNA-seq Experimental Design
| Replicate Type | Definition | Purpose | Example in Drug Discovery |
|---|---|---|---|
| Biological Replicates | Different biological samples or entities (e.g., individuals, animals, cells) | Assess biological variability and ensure findings are reliable and generalizable | 3 different animals or cell samples in each experimental group (treatment vs. control) |
| Technical Replicates | The same biological sample, measured multiple times | Assess and minimize technical variation (sequencing runs, lab workflows, environment) | 3 separate RNA sequencing experiments for the same RNA sample |
Biological replicates are considerably more important than technical replicates for robust drug discovery applications, with at least 3 biological replicates per condition typically recommended, though 4-8 replicates per sample group better cover most experimental requirements [13]. Several bioinformatics tools used in differential expression analysis require a minimum number of replicates for reliable data output, further emphasizing their importance [13].
Batch effects represent systematic, non-biological variations in data that arise from how samples are collected and processed. In large-scale drug discovery studies spanning multiple timepoints, sites, or involving thousands of samples, batch effects are expected and must be addressed [13]. Strategic plate layout during experimental setup can enable effective batch correction during computational analysis. Various batch correction techniques and software tools are available to remove these confounding effects [13]. Experimental controls, including artificial spike-in controls like SIRVs, are valuable tools that enable researchers to measure complete assay performance—particularly dynamic range, sensitivity, reproducibility, isoform detection, and quantification accuracy [13]. These controls provide internal standards for quantifying RNA levels between samples, normalizing data, assessing technical variability, and serving as quality control measures for large-scale experiments to ensure data consistency [13].
Bulk RNA-seq Methodologies and Protocols
Sample Preparation and Library Generation
The bulk RNA-seq workflow begins with sample preparation that varies depending on the source material—which can range from cultured cells and tissues to whole organisms [8] [7]. A critical first step involves converting RNA molecules into complementary DNA (cDNA) and preparing sequencing libraries compatible with next-generation sequencing platforms [7]. Because ribosomal RNA (rRNA) constitutes more than 80% of total RNA and is typically not the analytical focus, it is generally removed during sample preparation through either ribo-depletion or polyA-selection that enriches for messenger RNA (mRNA) [7].
Library preparation methods have evolved significantly, with choice of method depending on the specific drug discovery application:
Table 2: Bulk RNA-seq Library Preparation Methods for Drug Discovery
| Method Type | Key Features | Best Applications in Drug Discovery | Sample Input Considerations |
|---|---|---|---|
| Standard Full-Length (TruSeq, NEBNext) | Random primed cDNA, fragments entire transcript | Comprehensive transcriptome analysis, isoform detection | Higher input requirements, suitable for most sample types |
| 3'-Seq Methods (QuantSeq, LUTHOR) | Focus on 3' end, often extraction-free | Large-scale drug screens, gene expression and pathway analysis | Lower input, compatible with direct lysate protocols |
| Early Barcoding (Prime-seq) | Early sample barcoding, cost-efficient | High-throughput studies, large cohort analysis | Cost-effective for large sample numbers |
For large-scale drug screens based on cultured cells aimed at assessing gene expression patterns or pathways, 3'-Seq approaches with library preparation directly from lysates offer significant advantages by omitting RNA extraction, saving time and resources, and enabling efficient handling of larger sample numbers through early sample pooling [13]. When isoforms, fusions, non-coding RNAs, or variants are of interest, whole transcriptome approaches combined with mRNA enrichment or ribosomal rRNA depletion are preferable [13]. Specialized workflows exist for challenging sample types like whole blood or FFPE material, requiring careful extraction to remove contaminants, abundant transcripts (e.g., globin), genomic DNA, and to process low-quality and low-quantity samples [13].
Prime-seq represents a particularly efficient early barcoding bulk RNA-seq method that performs equivalently to standard TruSeq approaches but with substantially improved cost efficiency due to almost 50-fold cheaper library costs [23]. This method uses poly(A) priming, template switching, early barcoding, and Unique Molecular Identifiers (UMIs) to generate 3' tagged RNA-seq libraries, making it ideal for large-scale drug discovery applications requiring substantial sample numbers [23].
Comprehensive Workflow from Sample to Data
The complete bulk RNA-seq workflow encompasses multiple stages from sample preparation through to differential expression analysis, with quality control checkpoints at each step to ensure data integrity:
Two primary computational approaches exist for converting raw sequencing data into gene expression counts: alignment-based and pseudoalignment methods [6]. Alignment-based approaches using tools like STAR involve formal alignment of sequencing reads to either a genome or transcriptome, producing detailed alignment files that facilitate comprehensive quality checks but require substantial computational resources [6]. Pseudoalignment methods employed by tools like Salmon and kallisto use faster substring matching to probabilistically determine transcript origin without base-level alignment, offering significant speed advantages particularly valuable when processing thousands of samples [6]. A hybrid approach that uses STAR for initial alignment to generate QC metrics followed by Salmon for expression quantification leverages the strengths of both methods [6].
Data Analysis Frameworks for Drug Discovery
Quality Control and Data Exploration
Initial data exploration represents a critical first step in bulk RNA-seq analysis, assessing whether patterns in the raw data conform to the experimental design and identifying potential issues such as batch effects, outlier samples, or sample swaps [58]. Principal Component Analysis (PCA) serves as a primary method for visualizing variation within a dataset, reducing the number of gene "dimensions" to a minimal set of linearly transformed dimensions that reflect total variation [10]. The first principal component (PC1) describes the most variation within the data, PC2 the second most, and so forth, with the percentage of variation represented by each component calculable and visualizable through scree plots [10]. In well-designed drug discovery experiments, intergroup variability (differences between treatment and control conditions) should exceed intragroup variability (technical or biological variability between replicates) [10].
Quality control metrics should include assessments of read quality, alignment rates, genomic distribution of reads (exonic, intronic, intergenic), and sample-to-sample correlations. For drug discovery applications, particular attention should be paid to ensuring that control samples cluster together appropriately and that treatment groups show expected separation patterns consistent with the experimental design [58]. The data import process typically involves reading count data into R or Python environments, with tools like tximport used to bring in quantification files from tools like Salmon and summarize transcript-level counts to gene-level counts using transcript-to-gene mapping tables [58].
Differential Expression Analysis
Differential expression analysis identifies genes showing statistically significant changes in expression between experimental conditions—a fundamental task in drug discovery for identifying compound responses, mechanism of action, and biomarker discovery. The data analysis workflow typically involves:
Multiple software packages are available for differential expression analysis of bulk RNA-seq data, with DESeq2 and limma being among the most widely used [6] [59] [60]. These tools employ statistical models that account for the count-based nature of RNA-seq data and its inherent variability, with DESeq2 using a negative binomial generalized linear model specifically designed for RNA-seq count data [60]. The analysis typically produces measures of log2 fold change between conditions along with associated statistical significance values, which are then adjusted for multiple testing using methods like the Benjamini-Hochberg procedure to control false discovery rates [10].
Functional Interpretation and Pathway Analysis
Following identification of differentially expressed genes, functional interpretation through pathway and gene set enrichment analysis provides biological context to the transcriptional changes observed in drug treatment studies. Gene Ontology (GO) enrichment analysis identifies biological processes, molecular functions, and cellular compartments that are overrepresented among differentially expressed genes [10]. Additional pathway analysis methods like Gene Set Enrichment Analysis (GSEA) evaluate whether defined sets of genes (e.g., from KEGG, Reactome, or MSigDB) show statistically significant, concordant differences between experimental conditions, often revealing more subtle changes across related genes that might not reach individual significance thresholds but are biologically important [61].
For drug discovery applications, connection to known drug targets, disease pathways, and mechanism-of-action signatures enhances the translational relevance of findings. Integration with external databases linking genes to compounds, side effects, and clinical outcomes can further prioritize candidate genes and pathways for therapeutic development.
Applications in Drug Development Pipeline
Target Identification and Validation
Bulk RNA-seq provides a powerful approach for target identification during early drug discovery by comparing gene expression profiles between disease and healthy states, across different disease subtypes, or in response to genetic or chemical perturbations [8] [13]. By identifying consistently dysregulated genes and pathways in disease contexts, researchers can nominate potential therapeutic targets for further validation. The population-level perspective of bulk RNA-seq makes it particularly valuable for understanding overall pathway dysregulation and identifying master regulators of disease processes.
In the context of personalized medicine, bulk RNA-seq can identify expression signatures that define patient subgroups likely to respond to specific targeted therapies, enabling more precise patient stratification [13]. For example, transcriptional profiling of cancer samples has identified subtypes with distinct clinical outcomes and drug sensitivities, leading to more targeted therapeutic approaches. The use of both bulk and single-cell RNA-seq in tandem has proven particularly powerful, as demonstrated in a study of B-cell acute lymphoblastic leukemia (B-ALL) where both approaches were leveraged to identify developmental states driving resistance and sensitivity to the chemotherapeutic agent asparaginase [8].
Mechanism of Action Studies and Biomarker Discovery
Bulk RNA-seq plays a crucial role in elucidating mechanisms of drug action by characterizing transcriptomic changes following compound treatment [13]. Time-course experiments can distinguish primary drug targets from secondary adaptive responses, while dose-response studies establish relationships between compound exposure and transcriptional effects [13]. Kinetic RNA-seq approaches with methods like SLAMseq can globally monitor RNA synthesis and decay rates, providing dynamic information about transcriptional regulation that further refines understanding of drug mechanisms [13].
For biomarker discovery, bulk RNA-seq enables identification of gene expression signatures correlated with treatment response, disease progression, or patient outcomes [8] [13]. These signatures may include individual genes or sets of co-expressed genes that serve as predictive or prognostic indicators, potentially supporting patient selection for clinical trials or companion diagnostic development. The population-level resolution of bulk RNA-seq makes it particularly suitable for identifying robust biomarkers that generalize across patient populations, while its cost efficiency facilitates the larger sample sizes needed for biomarker validation studies.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Bulk RNA-seq in Drug Discovery
| Category | Specific Tools/Reagents | Function in Workflow | Application Notes |
|---|---|---|---|
| Library Prep Kits | TruSeq, NEBNext, Prime-seq | Convert RNA to sequencing-ready libraries | Prime-seq offers 4x cost efficiency via early barcoding [23] |
| RNA Quantification | Qubit, Bioanalyzer, TapeStation | Assess RNA quality and quantity | RIN >7.0 recommended for optimal library prep [10] |
| Spike-in Controls | ERCC, SIRVs | Normalization and QC standards | Essential for quality control in large studies [13] |
| Alignment Tools | STAR, HISAT2, Bowtie2 | Map sequencing reads to reference genome | STAR is splice-aware; preferred for genomic alignment [6] [59] |
| Quantification Tools | Salmon, kallisto, featureCounts | Generate expression counts | Salmon enables alignment-free quantification [6] [60] |
| Differential Expression | DESeq2, limma, edgeR | Identify statistically significant expression changes | DESeq2 uses negative binomial model for count data [6] [60] |
| Functional Analysis | clusterProfiler, GSEA, Enrichr | Pathway and gene set enrichment | Provides biological context to expression changes [10] [61] |
Bulk RNA sequencing remains an indispensable technology in the drug development pipeline, providing robust, quantitative transcriptomic data that accelerates therapeutic discovery and advances personalized medicine. When appropriately designed and executed, bulk RNA-seq experiments generate comprehensive gene expression profiles that identify novel drug targets, elucidate mechanisms of action, discover predictive biomarkers, and guide patient stratification strategies. The continuing evolution of bulk RNA-seq methodologies—including cost-efficient early barcoding approaches like Prime-seq and increasingly sophisticated analytical frameworks—ensures its ongoing relevance amid the growing availability of single-cell technologies. For drug discovery researchers, mastery of bulk RNA-seq experimental design, methodological considerations, and analytical approaches provides powerful capabilities for translating molecular insights into therapeutic advances.
Optimizing Bulk RNA-Seq: Best Practices for Robust and Reproducible Results
Bulk RNA sequencing (RNA-seq) has established itself as a fundamental tool in transcriptomic research, enabling genome-wide analysis of gene expression across cell populations and tissues. The reliability of conclusions drawn from these experiments, however, is profoundly influenced by experimental design, with sample size standing as a paramount consideration. Determining the appropriate sample size (N) for bulk RNA-seq experiments represents a critical balancing act between statistical rigor and practical constraints. Underpowered studies with insufficient replicates yield unreliable results characterized by false discoveries and inflated effect sizes, while excessively large studies may waste precious resources and violate ethical principles in animal research. Within the context of a broader thesis on bulk RNA sequencing methodology, this technical guide examines the evidence-based principles for sample size determination that enable researchers to optimize experimental designs for both reliability and feasibility.
The challenge of sample size determination stems from the inherent characteristics of RNA-seq data. Unlike microarray technology that generates continuous data, bulk RNA-seq produces count data that typically follows a negative binomial distribution due to its discrete nature and observed over-dispersion. This complex statistical foundation, combined with the high-dimensionality of transcriptomic data (thousands of genes measured simultaneously), creates a landscape where traditional power calculations often prove inadequate. Consequently, researchers must navigate multiple factors including biological variability, effect sizes of interest, technical noise, and resource limitations when designing experiments that can yield biologically meaningful insights.
Quantitative Evidence: Establishing Sample Size Benchmarks
Empirical Findings from Large-Scale Murine Studies
Recent large-scale empirical studies provide the most robust guidance for sample size determination in bulk RNA-seq experiments. A comprehensive 2025 investigation systematically analyzed sample size requirements using large cohorts (N=30) of genetically modified and wild-type mice, establishing gold-standard benchmarks for the field [62]. This research employed a down-sampling approach to evaluate how smaller sample sizes recapitulated findings from the full cohort, with key performance metrics summarized in Table 1.
Table 1: Performance Metrics Across Sample Sizes in Murine Bulk RNA-Seq Studies
| Sample Size (N) | False Discovery Rate (FDR) | Sensitivity | Recommendation Level |
|---|---|---|---|
| N ≤ 4 | 28-38% | <30% | Avoid - Highly misleading |
| N = 5 | 25-35% | 30-40% | Inadequate |
| N = 6-7 | <50% | >50% | Minimum requirement |
| N = 8-12 | <20% | 70-85% | Optimal range |
| N > 12 | <15% | >85% | Diminishing returns |
The data revealed that experiments with N=4 or fewer replicates produced highly misleading results, with false discovery rates reaching 28-38% across different tissues, while simultaneously failing to detect many genuinely differentially expressed genes (sensitivity below 30%) [62]. The variability in false discovery rates across trials was particularly pronounced at low sample sizes, with FDR ranging between 10-100% depending on which N=3 mice were selected for each genotype. This variability stabilized noticeably once sample size reached N=6 [62].
For a cutoff of 2-fold expression differences, the research established that N=6-7 mice represents the minimum threshold to consistently decrease false positive rates below 50% while achieving detection sensitivity above 50%. However, the most favorable balance between statistical performance and resource investment was found in the N=8-12 range, which provided significantly better recapitulation of the full experiment [62]. The principle of "more is always better" held true for both sensitivity and false discovery metrics, at least within the maximum sets of N=30 examined in these studies.
The Pitfalls of Underpowered Designs and Alternative Strategies
A common strategy to salvage underpowered experiments is to raise the fold-change threshold for declaring differential expression. However, evidence demonstrates that this approach is no substitute for adequate sample sizes. Raising fold-change cutoffs in underpowered experiments consistently inflates effect sizes (Type M errors, also known as the "winner's curse") and causes a substantial drop in detection sensitivity [62]. This phenomenon leads to a biased representation of biological reality, where only genes with extreme expression changes are detected while more subtle but potentially important changes are overlooked.
The negative consequences of underpowered mouse studies extend beyond individual experiments, representing a major factor driving the lack of reproducibility in the scientific literature [62]. The tension between statistical ideals and practical constraints is particularly acute in specialized research contexts, such as drug discovery, where sample availability may be limited. In such scenarios, consultation with bioinformaticians during the planning phase becomes essential to understand the statistical limitations and properly interpret results within these constraints [13].
Methodological Framework: Experimental Protocols for Sample Size Determination
Power Analysis Approaches for Bulk RNA-Seq
Statistical power in transcriptomic experiments refers to the probability of detecting true differentially expressed genes (DEGs), with sufficient power being essential for biologically meaningful findings [63]. Power analysis for bulk RNA-seq must account for several distinctive characteristics of this data type. The negative binomial distribution has gained popularity for modeling RNA-seq count data, as it effectively captures the over-dispersion commonly observed in sequencing experiments [63]. Tools such as 'edgeR', 'DESeq2', and 'baySeq' employ this distribution framework for DEG detection, while the 'voom' method applies normal-based theory to log-transformed count data [63].
When designing a power analysis for bulk RNA-seq, researchers must consider multiple factors that influence statistical power. These include the desired false discovery rate (FDR) threshold rather than type I error rate, given the simultaneous inference of thousands of genes; the effect sizes of biological interest; the biological variability inherent in the system under study; and the sequencing depth [63]. Numerous power analysis software tools have been developed to calculate required sample sizes, with model parameters often estimated from pilot data or provided stored datasets. However, researchers must exercise caution, as inappropriate use of stored data can lead to highly inaccurate sample size recommendations [63].
Table 2: Essential Components for Bulk RNA-Seq Power Analysis
| Component | Considerations | Tools/Approaches |
|---|---|---|
| Statistical Model | Over-dispersion, count data distribution | Negative binomial models, voom transformation |
| Effect Size | Biological relevance, fold-change thresholds | Based on pilot data or literature |
| Variability Estimation | Biological vs. technical variation, tissue type | Pilot studies, comparable published datasets |
| Error Control | Multiple testing correction, FDR threshold | Benjamini-Hochberg, target FDR typically 5-10% |
| Experimental Factors | Sequencing depth, replicate type | Trade-offs between replicates and depth |
The trade-off between biological replicates and sequencing depth represents another critical consideration in experimental design. Evidence demonstrates that the number of biological replicates has a greater influence on power than sequencing depth, particularly for detecting differentially expressed genes [63]. This principle should guide resource allocation decisions during experimental planning.
Empirical Evaluation Protocol Using Down-Sampling
The empirical protocol for establishing sample size requirements involves a down-sampling approach from large cohorts, which has proven highly informative for establishing field standards [62]. The following methodology outlines this approach:
Establish a Gold Standard: Conduct RNA-seq profiling on large cohorts (e.g., N=30 per condition) under carefully controlled conditions to minimize confounding variation. This large-N dataset serves as the benchmark representing the most accurate approximation of true biological effects [62].
Sub-Sampling Procedure: For each sample size N (typically ranging from 3 to 29), randomly sample N individuals from each condition without replacement. Repeat this process through multiple Monte Carlo trials (e.g., 40 iterations) to account for sampling variability [62].
Differential Expression Analysis: Perform DEG analysis on each sub-sampled dataset using standardized thresholds for statistical significance (e.g., adjusted p-value < 0.05) and fold-change (e.g., 1.5-fold) [62].
Performance Metric Calculation: For each sub-sampled signature, calculate sensitivity (percentage of gold standard genes detected) and false discovery rate (percentage of sub-sampled signature genes missing from the gold standard) [62].
Trend Analysis: Examine how both sensitivity and FDR change as functions of sample size, identifying points of diminishing returns and optimal ranges for balancing statistical performance with practical constraints [62].
This methodological framework can be adapted to various biological contexts and experimental conditions, providing researchers with a robust approach for establishing sample size requirements specific to their research system.
Implementation Guide: Practical Considerations for Experimental Design
Context-Specific Recommendations Across Research Domains
Practical sample size decisions must account for the specific research context and its associated constraints. While the empirical evidence provides general guidelines, implementation varies across research domains:
Basic Biological Research: For studies where sample availability is not limiting, such as cell lines or easily accessible model organisms, aiming for the optimal range of 8-12 biological replicates provides sufficient power for most applications. Pilot studies with 3-4 replicates can help estimate variability and refine sample size calculations for the full experiment [13].
Drug Discovery Applications: In pharmaceutical contexts, RNA-seq experiments serve various purposes throughout the development pipeline. For large-scale compound screening, where throughput is prioritized, smaller sample sizes may be acceptable for initial hits, with follow-up validation using larger cohorts. For mode-of-action studies, more robust sample sizes (6-8 replicates) are recommended to ensure reliable conclusions about drug effects [13].
Clinical and Biobank Studies: When working with precious human samples, such as patient biopsies or biobank specimens, practical limitations often restrict sample sizes. In these scenarios, researchers must acknowledge the statistical limitations of small sample sizes and employ complementary validation approaches. Strategic pooling of samples or utilization of public data resources for augmenting analyses may be considered [13].
The ENCODE consortium standards recommend a minimum of two biological replicates for bulk RNA-seq experiments, with higher replicates required for experiments with greater variability. Replicate concordance should achieve Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates [11].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Materials and Reagents for Bulk RNA-Seq Experiments
| Item | Function | Examples/Considerations |
|---|---|---|
| Spike-in Controls | Normalization, technical variability assessment | ERCC Spike-in Mix (Ambion), SIRVs; added at ~2% of final mapped reads [11] |
| RNA Preservation Reagents | RNA stabilization pre-extraction | RNAlater, TRIzol, PicoPure Extraction Buffer [10] |
| Library Prep Kits | cDNA library construction from RNA | NEBNext Ultra DNA Library Prep, 3'-seq methods for large screens [13] |
| mRNA Enrichment | Target selection | NEBNext Poly(A) mRNA magnetic isolation, rRNA depletion [10] |
| Quality Control Tools | RNA and library QC | Bioanalyzer, TapeStation (RIN >7.0 recommended) [10] |
| Reference Materials | Genome alignment and annotation | GENCODE annotations, STAR or TopHat indices [11] |
Strategic Planning for Resource-Limited Scenarios
When ideal sample sizes are not feasible due to resource constraints, researchers can employ several strategies to maximize the value of available resources:
Pilot Studies: Small-scale pilot experiments with 3-4 replicates provide invaluable data for estimating biological variability, which directly informs power calculations for larger studies. Pilots also allow optimization of experimental protocols before committing significant resources [13].
Leveraging Public Data Resources: Existing RNA-seq databases such as GEO, EMBL Expression Atlas, GTEx, and TCGA contain vast amounts of publicly available data that can be used to estimate parameters for power calculations or to augment experimental data through meta-analytical approaches [64].
Sequencing Depth Considerations: When biological replicates are limited, moderately increasing sequencing depth may provide some improvement in detection power for low-abundance transcripts, though this approach is less effective than adding biological replicates [63].
Robust Experimental Controls: Implementing rigorous experimental controls, including randomization, proper blinding, and careful batch design, becomes even more critical when sample sizes are limited, as it helps minimize confounding technical variation [10].
Visualizing Experimental Workflows and Relationships
Diagram 1: Sample size decision workflow
Diagram 1 illustrates a systematic decision framework for determining appropriate sample sizes in bulk RNA-seq experiments. This workflow incorporates key considerations such as sample availability constraints, expected effect sizes, and prior knowledge of variability, guiding researchers toward evidence-based sample size decisions that balance statistical power with practical limitations.
Diagram 2: Empirical methodology for establishing sample size requirements
Diagram 2 outlines the key methodological steps for empirically determining sample size requirements through down-sampling approaches. This three-phase process begins with establishing a gold standard using large cohorts, proceeds through systematic sub-sampling across multiple trials, and concludes with comprehensive performance evaluation to identify optimal sample size ranges.
Determining appropriate sample sizes for bulk RNA-seq experiments remains a critical challenge that balances statistical rigor with practical constraints. Empirical evidence establishes that very small sample sizes (N≤4) produce highly misleading results with inflated false discovery rates and poor sensitivity, while the range of N=8-12 represents an optimal balance for most experimental scenarios. Rather than relying on arbitrary rules or raising fold-change thresholds as substitutes for adequate replication, researchers should employ systematic approaches including power analyses, pilot studies, and strategic utilization of public data resources. By adopting these evidence-based guidelines and maintaining transparency about statistical limitations, the research community can enhance the reliability and reproducibility of transcriptomic findings while making responsible use of precious scientific resources.
Batch effects are systematic technical variations introduced during the handling and processing of samples that are unrelated to the biological factors of interest. In bulk RNA sequencing (RNA-seq), these non-biological variations can compromise data reliability, obscure true biological differences, and significantly reduce the statistical power to detect genuinely differentially expressed (DE) genes [65] [66]. The profound negative impact of batch effects extends beyond mere data noise; they represent a paramount factor contributing to irreproducibility in omics studies, potentially leading to retracted articles, discredited research findings, and substantial economic losses [66]. In clinical settings, batch effects have even resulted in incorrect classification outcomes for patients, directly affecting treatment decisions [66].
The fundamental cause of batch effects in RNA-seq data can be partially attributed to the basic assumptions of data representation. The relationship between the actual abundance of an analyte in a sample and the instrument readout is assumed to be linear and fixed. However, in practice, this relationship fluctuates due to differences in diverse experimental factors, making intensity measurements inherently inconsistent across different batches [66]. Understanding, mitigating, and correcting for these effects is therefore crucial for ensuring the reliability and reproducibility of RNA-seq data throughout the drug discovery pipeline, from target identification to mode-of-action studies [13].
Experimental Design: The First Line of Defense
Strategic experimental design represents the most effective and proactive approach to minimizing batch effects. A well-designed experiment can prevent many batch effect issues that are difficult or impossible to fully resolve through computational correction alone.
Batch effects can emerge at virtually every step of the RNA-seq workflow. The table below categorizes common sources and corresponding mitigation strategies.
Table 1: Common Sources of Batch Effects and Proactive Mitigation Strategies
| Experimental Phase | Source of Batch Effects | Mitigation Strategy |
|---|---|---|
| Study Design | Confounded design (batch correlated with condition) | Randomize samples across batches; ensure each batch contains representatives of all experimental conditions [27] [66] |
| Insufficient replicates | Include at least 3-4 biological replicates per condition [27] [67] | |
| Sample Preparation | Different RNA isolation days/operators | Perform all RNA isolations simultaneously by a single operator [67] [10] |
| Different reagents/library prep kits | Use the same reagent lots and kits for all samples [27] | |
| Sequencing | Different sequencing runs/lanes | Multiplex all samples together and run on the same lane; if impossible, balance conditions across lanes [67] |
| Different sequencing depths | Use consistent library preparation protocols and sequencing depths [27] |
Strategic Replication and Randomization
Biological replicates—independent biological samples of the same condition—are absolutely essential for differential expression analysis as they allow for the measurement of biological variation between samples [27]. While technical replicates (repeated measurements of the same biological sample) were once common in microarray studies, they are generally unnecessary with modern RNA-seq technologies where technical variation is much lower than biological variation [27].
The number of replicates has a direct impact on statistical power. As a best practice, a minimum of 3 biological replicates per condition is recommended, with 4 being the optimum minimum for most applications [67]. For cell line studies, biological replicates should be "performed as independently as possible," meaning that cell culture media should be prepared freshly for each experiment, and different frozen cell stocks and growth factor batches should be used [27].
Randomization is equally critical. To avoid confounding, animals in each condition should ideally be of the same sex, age, litter, and batch. If this is not possible, animals must be split equally between conditions [27]. For example, if using both male and female animals, each experimental group should contain a similar proportion of males and females rather than having all controls be female and all treatments be male.
Plate and Batch Layout Design
In large-scale studies where processing all samples simultaneously is impossible, careful batch design is essential. The fundamental rule is: do NOT confound your experiment by batch [27]. Instead, replicates of the different sample groups must be split across batches.
For instance, in an experiment with three treatment groups (A, B, C) and four replicates per group, where only two samples can be processed at a time, the six processing batches should each contain samples from different treatment groups rather than grouping all replicates of the same treatment together [27]. This balanced design ensures that batch effects can be measured and removed bioinformatically without being confounded with the biological effects of interest.
Sample Processing and Technical Controls
Standardization of Laboratory Protocols
Consistency in sample processing is paramount for minimizing technical variation. All RNA extractions should be performed at the same time by the same operator using the same reagents to prevent introduction of batch effects [67] [10]. Similarly, library preparations for all samples should be conducted simultaneously using the same reagent lots and protocols. Any deviation from this principle introduces variability that can manifest as batch effects in downstream analyses.
For cell line experiments specifically, it is recommended that preparation across all conditions be performed at the same time, even while maintaining independent biological replicates [27]. This balances the need for technical consistency with biological independence.
Utilization of Spike-In Controls
Artificial spike-in controls, such as SIRVs (Spike-In RNA Variants), are valuable tools in RNA-seq experiments that enable researchers to measure the performance of the complete assay [13]. These commercially developed RNA sequences are added in known quantities to each sample before library preparation and serve multiple functions:
- Provide an internal standard for quantifying RNA levels between samples
- Enable assessment of technical variability across batches
- Help normalize data and evaluate quantification accuracy
- Serve as quality control measures for large-scale experiments to ensure data consistency
Spike-in controls are particularly useful in large-scale drug discovery screens where samples may be processed in multiple batches over time, as they provide an objective metric for tracking technical performance across batches [13].
Table 2: Essential Research Reagent Solutions for Batch Effect Mitigation
| Reagent/Solution | Function in Batch Effect Mitigation |
|---|---|
| Spike-In Controls (e.g., SIRVs) | Internal standards for normalization and quality control across batches [13] |
| Consistent RNA Extraction Kits | Minimizes technical variation from sample preparation [27] [67] |
| Single-Lot Library Prep Kits | Reduces batch effects introduced during library construction [27] |
| Uniform Sequencing Kits | Ensures consistent sequencing chemistry across all samples [67] |
Computational Correction Methods
When batch effects cannot be avoided through experimental design, computational correction methods offer a powerful solution for mitigating their impact during data analysis.
Batch Effect Detection and Diagnosis
Before applying any correction method, it is crucial to detect and diagnose batch effects in the data. Principal Component Analysis (PCA) is the most common visualization tool for this purpose. In a PCA plot, samples typically cluster by biological group in a well-controlled experiment. However, when batch effects are present, samples may instead cluster by processing date, sequencing lane, or other technical factors [27] [19].
The figure below illustrates the impact of proper experimental design and batch effect correction on sample clustering in PCA plots.
Figure 1: Impact of Experimental Design on Batch Effect Detection and Correction
Several computational approaches exist for correcting batch effects in RNA-seq data, each with different underlying assumptions and methodologies:
Covariate Adjustment: Methods like those implemented in edgeR and DESeq2 include batch as a covariate in the linear model during differential expression analysis [19] [65]. This approach accounts for batch effects but does not return a corrected count matrix.
Empirical Bayes Methods: ComBat-seq uses an empirical Bayes framework with a negative binomial model to adjust count data directly, preserving the integer nature of RNA-seq counts while removing batch effects [65].
Reference-Based Correction: ComBat-ref, a refinement of ComBat-seq, selects the batch with the smallest dispersion as a reference and adjusts other batches toward this reference, demonstrating superior performance in maintaining statistical power while effectively mitigating batch effects [68] [65].
Machine Learning Approaches: Recently, machine learning methods have been proposed to address batch effects by modeling discrepancies among batches, though these can carry higher risks of over-correction if not carefully implemented [65] [69].
Table 3: Comparison of Batch Effect Correction Methods for RNA-seq Data
| Method | Underlying Model | Key Feature | Preserves Count Integrity | Best For |
|---|---|---|---|---|
| DESeq2/edgeR Covariate | Negative Binomial GLM | Includes batch as covariate in DE model | Yes | Standard DE analysis with simple batch structure [19] [65] |
| ComBat-seq | Negative Binomial + Empirical Bayes | Directly adjusts count data | Yes | Complex batch effects across multiple batches [65] |
| ComBat-ref | Negative Binomial + Reference Batch | Adjusts batches toward low-dispersion reference | Yes | Scenarios with varying dispersion between batches [68] [65] |
| NPMatch | Nearest-Neighbor Matching | Matches samples across batches | Varies | Datasets with many biological replicates [65] |
The ComBat-ref Workflow
ComBat-ref has demonstrated particularly strong performance in simulations and real datasets. The method follows this workflow:
- Model RNA-seq counts using a negative binomial distribution, allowing each batch to have different dispersions
- Estimate batch-specific dispersion parameters by pooling gene count data within each batch
- Select the reference batch with the smallest dispersion
- Adjust other batches toward the reference batch using a generalized linear model (GLM) framework
- Preserve count data for the reference batch while adjusting counts in other batches through cumulative distribution function (CDF) matching [65]
In performance evaluations, ComBat-ref maintained exceptionally high statistical power—comparable to data without batch effects—even when there was significant variance in batch dispersions, and outperformed other methods when false discovery rate (FDR) was used for differential expression analysis [65].
Integrated Analysis Workflow
A comprehensive approach to batch effect management spans the entire RNA-seq workflow, from experimental design to final data interpretation. The following diagram outlines this integrated strategy.
Figure 2: Integrated Workflow for Batch Effect Management
Validation and Quality Assurance
After applying batch effect correction, validation is essential to ensure that technical artifacts have been removed without eliminating genuine biological signal. Several approaches can be used:
- PCA Visualization: Re-examine PCA plots after correction to verify that samples now cluster by biological condition rather than batch [19] [10].
- Known Positive Controls: Check whether expected differentially expressed genes (based on prior knowledge or positive controls) remain significant after correction.
- Negative Controls: Verify that genes known to be unaffected by the experimental conditions do not show spurious differential expression.
- Statistical Metrics: Evaluate the effectiveness of correction using metrics like the Silhouette score or other cluster validation measures.
It is also crucial to maintain awareness of the risk of over-correction, where true biological variation is inadvertently removed along with technical noise. This is particularly problematic when batch effects are subtle or when biological groups are partially confounded with batches [69] [66].
Batch effects remain a significant challenge in bulk RNA-seq experiments, particularly in large-scale drug discovery applications where samples must be processed in multiple batches. However, through careful experimental design that includes adequate replication, balanced batch layouts, and standardized protocols, many batch effects can be prevented or minimized. When unavoidable, computational correction methods like ComBat-ref provide powerful approaches for mitigating batch effects while preserving biological signal.
The field continues to evolve, with emerging methodologies including machine learning approaches and improved multi-omics integration techniques showing promise for more sophisticated batch effect management [65] [66]. Nevertheless, the foundation of effective batch effect control remains strategic experimental design—a principle that cannot be replaced by computational correction alone. By implementing the comprehensive strategies outlined in this guide, researchers can significantly enhance the reliability, reproducibility, and biological validity of their RNA-seq findings throughout the drug discovery and development pipeline.
Within the framework of a broader thesis on how bulk RNA sequencing works, quality control (QC) stands as the foundational pillar ensuring the validity and reproducibility of research findings. Bulk RNA sequencing provides a global snapshot of gene expression by measuring the average transcript levels across populations of thousands to millions of cells [24]. The reliability of this snapshot, however, is entirely dependent on the quality of the starting biological material and the technical execution of the sequencing workflow. This guide details the essential QC checkpoints, from initial RNA integrity assessment to post-sequencing metric evaluation, providing researchers, scientists, and drug development professionals with the protocols and standards necessary to generate robust and interpretable data.
Pre-sequencing Quality Control: RNA Integrity
The integrity of the input RNA is the most critical pre-analytical factor determining the success of a bulk RNA-seq experiment. Degraded RNA can lead to biased gene expression estimates, particularly for longer transcripts, compromising all subsequent analyses [70].
RNA Integrity Number (RIN)
The gold standard for assessing RNA quality is the RNA Integrity Number (RIN), an algorithm developed by Agilent Technologies that assigns an integrity value from 1 (completely degraded) to 10 (perfectly intact) [70] [71].
- Principle: The RIN algorithm uses capillary electrophoresis to generate an electropherogram trace of the RNA sample. It goes beyond the traditional 28S/18S rRNA ratio by incorporating multiple features from this trace, including the total RNA ratio, the height of the 28S peak, and the signal in the fast region (degradation products) [70] [71].
- Interpretation and Thresholds: The following table summarizes the general interpretation of RIN scores and their suitability for different downstream applications. However, it is crucial to validate what constitutes an acceptable RIN for your specific sample type and experimental question [71].
Table: Interpretation of RNA Integrity Number (RIN) Scores
| RIN Score | Integrity Level | Suitability for Downstream Applications |
|---|---|---|
| 8 - 10 | High Integrity | Ideal for most applications, including RNA-Seq and microarrays [71]. |
| 7 - 8 | Moderate Integrity | Generally acceptable for gene arrays and qPCR [71]. |
| 5 - 6 | Low Integrity | Marginal; may be suitable for RT-qPCR but requires caution [71]. |
| 1 - 5 | Severe Degradation | Unsuitable for most gene expression studies [71]. |
- Limitations: Researchers must be aware of the limitations of RIN. It primarily reflects the integrity of ribosomal RNAs, which may not always correlate perfectly with the stability of messenger RNAs (mRNAs) or other biomarkers of interest [70]. Furthermore, the standard RIN algorithm is designed for mammalian RNA and may perform poorly with plant samples or in studies involving eukaryotic-prokaryotic interactions due to its inability to differentiate between different types of ribosomal RNA [70].
Pre-sequencing Laboratory Protocol
A typical workflow for preparing and qualifying a bulk RNA-seq library is as follows, incorporating key QC checkpoints.
Figure 1: Bulk RNA-seq laboratory workflow with key QC checkpoints.
Detailed Methodologies:
- RNA Isolation & QC1: RNA Integrity Check: RNA is isolated from the bulk sample (e.g., tissue, sorted cells) using a protocol that effectively inactivates RNases. The resulting RNA is quantified using a method like Qubit, and its integrity is assessed, typically on an Agilent Bioanalyzer or TapeStation system to generate a RIN. A RIN of >7.0 is often used as a minimum threshold, with >8.0 being ideal for RNA-seq [10] [71]. This step is critical for determining the sample's suitability for proceeding.
- Library Preparation: The high-quality RNA undergoes library preparation. This typically involves selecting for mRNA via poly(A) capture, followed by reverse transcription into cDNA, adapter ligation, and PCR amplification [10] [24]. Alternatively, ribodepletion can be used to remove abundant rRNA. Some modern protocols like prime-seq use early barcoding and template switching for increased cost efficiency [23].
- QC2: Library Quality Check: The final cDNA library is quality-controlled again before sequencing. This involves checking the library's concentration (e.g., with Qubit) and its size distribution (e.g., with an Agilent TapeStation) to ensure adapter ligation was successful and that there is no adapter dimer contamination [24]. Normalizing samples to the same concentration at this stage helps minimize read count variability during sequencing [24].
Post-sequencing Quality Control: Sequencing Metrics
After sequencing, raw data must be processed and evaluated to ensure the sequencing run itself was successful and that the data is of high quality for downstream analysis. The primary steps involve quality assessment, read alignment, and quantification.
Figure 2: Bioinformatic processing and QC workflow for bulk RNA-seq data.
Essential Sequencing Metrics and Their Interpretation
The following table outlines key post-sequencing QC metrics, their ideal values, and the potential implications of deviations.
Table: Essential Post-Sequencing QC Metrics for Bulk RNA-seq
| Metric | Description & Ideal Value | Potential Issue if Metric Fails |
|---|---|---|
| Total Reads | The total number of raw sequencing reads. Sufficient depth (e.g., 20-50 million per sample) is needed for accurate quantification [72]. | Underloading/overloading of sequencer; insufficient statistical power to detect differentially expressed genes. |
| % rRNA Reads | Percentage of reads mapping to ribosomal RNA. Should be low (e.g., 4-10%), depending on the rRNA removal method used [72]. | Inefficient ribodepletion or poly(A) selection, wasting sequencing reads on uninformative rRNA. |
| Mapping Rate | Percentage of reads that successfully align to the reference genome. Ideally >80% [72] [23]. | High levels of contamination, poor RNA quality, or use of an incorrect reference genome. |
| Exonic/Intronic Rate | The proportion of reads mapping to exons vs. introns. Poly(A)-selected libraries should have a high exonic rate; ribodepleted will have more intronic reads [72]. | DNA contamination if intronic rate is unexpectedly high in poly(A)-selected libraries (though ribodepleted libraries naturally have more intronic reads from nascent RNA) [23]. |
| Genes Detected | The number of unique genes with non-zero expression. Indicates library complexity [72]. | Low complexity can result from technical issues (degradation, poor amplification) or biologically low-diversity samples. |
| Duplicate Reads | Reads mapping to the exact same genomic location. High levels can indicate PCR over-amplification [72]. | In RNA-seq, some duplicates are expected from highly expressed genes; specialized tools are used to differentiate technical from biological duplicates [72]. |
Post-sequencing Analysis Protocol
The standard bioinformatic processing and QC pipeline involves the following steps, often automated using workflows like the nf-core/rnaseq pipeline [6]:
- Quality Control & Read Trimming (FastQC, Trimmomatic): Raw FASTQ files are first assessed with tools like FastQC to evaluate per-base sequence quality, GC content, adapter contamination, and overrepresented sequences. Based on this, tools like Trimmomatic or the FASTX-Toolkit are used to trim low-quality bases and adapter sequences [19] [73].
- Read Alignment (STAR): The cleaned reads are aligned to a reference genome using a splice-aware aligner such as STAR. This step accounts for introns in eukaryotic transcripts [19] [6].
- Gene Quantification (HTSeq-count, Salmon): The aligned reads are assigned to genomic features (genes) using tools like HTSeq-count to generate a count matrix—a table where rows represent genes, columns represent samples, and values are the number of reads assigned to each gene [19]. Alternatively, alignment-free tools like Salmon can be used for fast and accurate quantification [6].
- Post-Alignment QC: The output of the alignment and quantification steps is used to calculate the metrics described in the table above (e.g., mapping rate, % rRNA, genes detected). This is a critical checkpoint before proceeding to statistical analysis.
The Scientist's Toolkit: Essential Reagents and Tools
Table: Key Research Reagent Solutions and Software for Bulk RNA-seq QC
| Item | Function |
|---|---|
| Agilent Bioanalyzer/TapeStation | Instrument systems that perform capillary electrophoresis to assess RNA quality and quantity, generating the RIN [70] [71]. |
| Qubit Fluorometer | Instrument for highly accurate nucleic acid quantification using fluorescent dyes, superior to spectrophotometry for library quantification [24]. |
| Poly(A) Selection or Ribo-depletion Kits | Reagent kits to enrich for mRNA by capturing polyadenylated transcripts or by removing abundant ribosomal RNA, thus increasing informative reads [72] [10]. |
| Trimmomatic | Software tool used to trim adapter sequences and remove low-quality bases from raw FASTQ files, improving overall data quality [19] [73]. |
| STAR Aligner | A widely used, splice-aware aligner for mapping RNA-seq reads to a reference genome [19] [6]. |
| HTSeq-count / featureCounts | Software packages that take aligned reads and a genome annotation file to generate a count matrix by assigning reads to genes [19]. |
| FastQC | A quality control tool that provides an overview of potential issues in raw sequencing data via an HTML report [73]. |
| DESeq2 / limma | R/Bioconductor packages for differential expression analysis that take the count matrix as input and apply statistical models to identify significantly changed genes [19] [6]. |
Rigorous quality control is non-negotiable in bulk RNA sequencing. It begins with the foundational assessment of RNA integrity via RIN and continues through multiple checkpoints in library preparation and post-sequencing bioinformatic analysis. By systematically evaluating metrics such as rRNA residue, mapping rates, and library complexity, researchers can safeguard their investment in sequencing, ensure the biological validity of their results, and draw meaningful conclusions in their research and drug development projects. Adhering to these outlined protocols and standards provides a robust framework for generating high-quality, reproducible transcriptomic data.
Bulk RNA Sequencing (RNA-Seq) is a powerful technique for assessing RNA expression in cells, enabling researchers to profile gene expression, identify novel RNA species, and analyze alternative splicing across entire transcriptomes [74]. A critical acknowledgment in any RNA-seq experiment is that the technique does not count absolute numbers of RNA copies in a sample; rather, it yields relative expression levels within the sampled RNA [74]. This relative nature makes the data particularly susceptible to technical variation, which arises not from biological differences but from the experimental process itself. These technical artifacts can originate from multiple sources, including library preparation, sequencing depth, and RNA quality, potentially obscuring true biological signals and leading to erroneous conclusions in downstream analysis.
The process begins with converting RNA into complementary DNA (cDNA), followed by adapter ligation, library amplification, and high-throughput sequencing [10]. At each step, technical variability can be introduced. For instance, library preparation protocols differ significantly in their handling of ribosomal RNA, their strandedness, and their efficiency, all of which systematically alter the resulting data [74]. Furthermore, differences in sequencing depth—the total number of reads sequenced per sample—can create apparent expression differences that are purely technical [27]. Even before sequencing, the quality of the input RNA, often measured by the RNA Integrity Number (RIN), profoundly affects the results, with degraded RNA biasing detection against longer transcripts [74]. Normalization methods are therefore not merely a preprocessing step but a fundamental statistical correction that is essential for distinguishing true biological differential expression from technical artifacts.
Understanding the specific sources of technical variation is a prerequisite for selecting appropriate normalization strategies and designing robust experiments.
Library Preparation and RNA Quality
The initial steps of RNA extraction and library construction are fertile ground for technical variation. The RNA integrity is paramount; degraded RNA, often indicated by a low RIN value, can severely skew expression measurements. Protocols that rely on poly(A) selection for mRNA enrichment perform poorly with degraded RNA because they require an intact polyA tail. In such cases, methods utilizing ribosomal RNA (rRNA) depletion combined with random priming are preferable, though they introduce their own variability [74]. The choice between stranded and unstranded library protocols also contributes to technical differences. Stranded libraries, which preserve the information about which DNA strand was transcribed, are preferred for accurately determining transcript orientation and analyzing non-coding RNAs, but they are often more complex and costly than unstranded alternatives [74]. A significant source of variation in library prep is the handling of ribosomal RNA, which constitutes approximately 80% of cellular RNA. Depletion strategies (e.g., using RNAseH or precipitating beads) are employed to reduce rRNA reads, but their efficiency and potential for off-target effects on non-ribosomal genes are variable and must be assessed for the specific genes of interest in a study [74].
Batch Effects
Batch effects represent a significant and pervasive source of technical variation in RNA-seq analyses. A batch effect occurs when samples are processed in different groups (batches) under slightly different conditions, and the technical differences between these batches can have a larger effect on the gene expression data than the actual biological variables of interest [27]. Batch effects can be introduced at multiple stages:
- RNA Isolation: Performing RNA extractions on different days or by different researchers [27].
- Library Preparation: Creating sequencing libraries in separate batches over time [10].
- Sequencing Run: Sequencing control and experimental samples on different flow cells or at different times [10].
The experimental design is the first line of defense against confounding by batch effects. The best practice is to avoid confounding by ensuring that replicates from each experimental condition are distributed across all batches. For example, if an experiment has three treatment groups (A, B, and C) and RNA can only be isolated from two samples at a time, the isolation batches should each contain a mix of samples from groups A, B, and C rather than containing only one group. This design allows the statistical model to later separate and adjust for the variation due to batch, provided that batch information is meticulously recorded in the experimental metadata [27].
Sequencing Depth and Replicate Design
Sequencing depth and the number of biological replicates are two key experimental design choices that are often in tension due to cost constraints. Sequencing depth refers to the total number of reads sequenced per sample. While deeper sequencing allows for better detection of lowly-expressed genes, evidence suggests that investing in more biological replicates generally provides greater statistical power for identifying differentially expressed genes than increasing sequencing depth [27]. The relationship between replicates, depth, and power is illustrated in a study where an increase in the number of replicates returned more differentially expressed genes than a comparable increase in sequencing depth [27]. General guidelines suggest that for gene-level differential expression, 15 million single-end reads per sample is often sufficient if there are a good number of replicates (more than three). For projects focused on detecting lowly-expressed genes or performing isoform-level differential expression, a depth of 30-60 million reads is recommended [27].
Core Normalization Methods: Theory and Application
Normalization aims to remove systematic technical variation, enabling valid comparisons of gene expression across samples. The following section details the most critical methods used in the field.
Foundational Normalization Strategies
Table 1: Foundational Normalization Methods for Bulk RNA-Seq Data
| Method | Core Principle | Key Assumptions | Best Suited For | Potential Limitations |
|---|---|---|---|---|
| Counts per Million (CPM) | Scales raw counts by the total number of reads per sample and a factor of one million. | The total number of reads per sample (library size) is the primary source of technical variation. | Initial data exploration; single-sample analysis. | Highly sensitive to expression of a few very abundant genes; not suitable for between-sample DE analysis. |
| Trimmed Mean of M-values (TMM) | Identifies a set of stable genes between a sample and a reference, trims extreme log fold-changes and library sizes, and uses the weighted mean of the remaining log fold-changes as a scaling factor. | Most genes are not differentially expressed, and those that are DE are symmetrically up- and down-regulated. | Comparisons between samples where the majority of genes are expected to be non-DE; implemented in edgeR. |
Performance can degrade with extreme, global expression changes or when the stable gene assumption is violated. |
| Relative Log Expression (RLE) | Calculates a scaling factor for each sample based on the median ratio of its counts to the geometric mean of counts across all samples. | The size factors are representative of technical differences, and most genes are non-DE. | Standard for many experiments; the default method in DESeq2. |
Assumes that the majority of the genome is not differentially expressed. Can be biased by large-scale differential expression. |
| Upper Quartile (UQ) | Scales counts based on the 75th percentile of counts, ignoring genes with zero counts. | The upper quartile of expression is stable across samples. | An alternative to total count normalization when a few highly expressed genes dominate the library size. | Less stable than TMM or RLE if the upper quartile itself is composed of differentially expressed genes. |
| Transcripts Per Million (TPM) | Normalizes for both sequencing depth and gene length. Reads are first normalized per kilobase of gene length, then per million of these scaled reads. | Allows for comparison of expression levels both within and between samples. | Comparing the relative abundance of different transcripts within a single sample. | Not designed for direct use in differential expression analysis between samples without further library size normalization. |
Workflow Integration of Normalization
Normalization is not a standalone step but is deeply integrated into the differential expression analysis workflow. The following diagram illustrates its critical position after data preprocessing and before statistical testing.
Diagram 1: The role of normalization in the RNA-Seq analysis workflow. After raw reads are processed and aligned to generate a count matrix, normalization corrects for technical variation before statistical testing for differential expression.
Advanced Considerations and Method Selection
The choice of normalization method can significantly impact the results of a study. For standard differential gene expression analysis, RLE (used in DESeq2) and TMM (used in edgeR) are the most widely adopted and robust methods. These methods are incorporated into established statistical frameworks that model count data using a negative binomial distribution to account for both technical variation and biological variability [10] [75]. For example, in a study of Post COVID-19 condition (PCC), count data were normalized using DESeq2 as a fundamental step before applying the generalized linear model for differential expression testing [75].
It is critical to understand that no single normalization method is universally superior. The optimal choice can depend on the specific characteristics of the dataset, such as the presence of extreme outliers, the symmetry of differential expression, or the fraction of genes that are truly differentially expressed. For complex projects, some researchers perform normalization using multiple methods and assess the consistency of the key results across them. Furthermore, when a study includes known batch effects, the design formula in tools like DESeq2 should include both the batch and the condition of interest to regress out the batch variation during the model fitting process [27] [75].
Experimental Protocols for Robust Normalization
Proper normalization rests on a foundation of sound experimental design and execution. The following protocols are essential for generating data where normalization can effectively address technical variation.
Protocol for a Bulk RNA-Seq Experiment with Batch Effect Control
This protocol outlines the key steps for a bulk RNA-seq study from tissue to data, with an emphasis on minimizing and controlling for batch effects.
Experimental Design and Sample Randomization:
- Determine the required number of biological replicates (aim for a minimum of 3, but more are always better) based on power considerations [27].
- Identify all potential batch effects (e.g., different days for RNA isolation, multiple library preparation operators, separate sequencing runs).
- Create a sample metadata table that explicitly records these factors. Crucially, randomize or block the processing order so that samples from all experimental groups are distributed across all batches [27]. For instance, if RNA is isolated in six batches for 12 samples from three groups, each batch should contain a representative mix of the groups.
RNA Extraction and Quality Control:
- Extract total RNA using a standardized, reproducible method. For blood samples, use RNA-stabilizing reagents like PAXgene tubes [75] [74].
- Perform rigorous QC on the extracted RNA. Use an instrument like an Agilent Bioanalyzer to determine the RNA Integrity Number (RIN). A RIN greater than 7 is generally considered acceptable for high-quality sequencing [74]. Also, check for contamination using Nanodrop 260/280 and 260/230 ratios.
Library Preparation with Balanced Batches:
- Select a library protocol appropriate for the research question (e.g., stranded vs. unstranded, polyA-selection vs. rRNA depletion) [74].
- Prepare sequencing libraries. If multiple library prep batches are unavoidable, ensure that each batch contains an equal representation of samples from all experimental conditions. Record the batch ID for each library in the metadata.
Sequencing and Data Generation:
- Sequence all libraries. Ideally, all samples should be run in a single sequencing lane to avoid a major batch effect. If multiple lanes are required, use a balanced design where each lane contains a mix of all sample groups.
- Demultiplex the raw sequencing data to generate FASTQ files.
Computational Analysis and Normalization:
- Perform quality control on the FASTQ files using tools like FastQC.
- Align the reads to the appropriate reference genome (e.g., GRCh38 for human) using a splice-aware aligner like STAR or HiSAT2.
- Generate a raw count matrix of reads mapped to each gene using tools like HTSeq or featureCounts.
- Input the raw count matrix and the sample metadata (including batch information) into a statistical environment like R.
- Normalize the data and account for batch effects using a established software package. For example, in DESeq2, this is achieved by using a design formula such as
~ batch + conditionduring the model fitting step, which normalizes the counts and adjusts for the specified batch effect during differential expression testing [75].
Protocol for Differential Expression Analysis with DESeq2
This protocol provides a detailed methodology for a typical differential expression analysis, with normalization as a core, automated step within the DESeq2 framework.
Data Input and Object Creation:
- Load the raw count matrix and sample metadata (colData) into R.
- Create a DESeqDataSet object using the
DESeqDataSetFromMatrix()function. Specify the design formula that includes the condition of interest and any known batch variables (e.g.,~ batch + group).
Pre-filtering (Optional but Recommended):
- Remove genes with very low counts across all samples to reduce the multiple testing burden and improve speed. A common threshold is to keep genes with at least 10 reads in total.
Normalization and Model Fitting:
- Execute the core analysis with the
DESeq()function. This single function call performs a multi-step process: a. Estimation of size factors (using the RLE method) to control for differences in library sizes. b. Estimation of dispersion for each gene. c. Fitting of a negative binomial generalized linear model and Wald statistics for hypothesis testing. - The normalization (size factor estimation) is performed internally and automatically.
- Execute the core analysis with the
Results Extraction and Exploration:
- Extract the results table of differentially expressed genes using the
results()function. Specify the contrast of interest (e.g.,contrast = c("group", "Treatment", "Control")). - The resulting log2 fold changes and p-values are based on the normalized count data, with technical variation accounted for by the size factors and any batch terms in the design.
- Extract the results table of differentially expressed genes using the
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Bulk RNA-Seq
| Item | Function/Application | Example Products/Tools |
|---|---|---|
| RNA Stabilization Reagent | Preserves RNA integrity immediately upon sample collection, preventing degradation that introduces technical variation. | PAXgene Blood RNA Tubes [75] [74] |
| RNA Extraction Kit | Isolates high-quality total RNA from biological samples. The consistency of extraction is critical for minimizing batch effects. | PicoPure RNA Isolation Kit, QIAsymphony PAXgene Blood RNA Kit [10] [75] |
| RNA Quality Assessment | Evaluates RNA integrity (RIN) and purity (260/280 ratio) to ensure only high-quality samples proceed to library prep. | Agilent 2100 Bioanalyzer, Agilent TapeStation [10] [75] [74] |
| Library Prep Kit | Converts RNA into a sequencing-ready library. Choice depends on required strandedness, input amount, and RNA quality. | NEBNext Ultra DNA Library Prep Kit, Illumina Stranded mRNA Prep [10] [74] |
| rRNA Depletion Kit | Selectively removes abundant ribosomal RNA, increasing the sequencing depth of informative mRNA and non-coding RNA. | RNAse H-based probes, Ribozero rRNA Removal Kit [74] |
| Analysis Software & Pipelines | Performs read alignment, count quantification, normalization, and differential expression analysis. | DESeq2, edgeR, inDAGO (GUI for non-bioinformaticians) [75] [76] |
| Reference Genome & Annotation | The genomic coordinate system for aligning reads and assigning them to genes. Essential for generating the count matrix. | GENCODE, Ensembl (e.g., GRCh38 for human, mm10 for mouse) [10] [75] |
Normalization is an indispensable component of bulk RNA-seq data analysis, serving as the statistical bulwark against confounding technical variation. The effectiveness of any normalization method, from established approaches like RLE and TMM to more complex batch correction models, is profoundly dependent on rigorous experimental design. This includes the strategic use of biological replicates, proactive avoidance of confounding, and meticulous documentation of batch information. As the application of bulk RNA-seq expands from basic research into clinical realms, such as the characterization of immune landscapes in myeloproliferative neoplasms and Post COVID-19 condition, the demand for robust, transparent, and well-understood normalization practices only intensifies [75] [77]. By systematically addressing technical variation through both sound laboratory practice and sophisticated computational correction, researchers can ensure that the biological signals they uncover are both accurate and meaningful.
Bulk RNA sequencing (RNA-Seq) is a foundational technique in molecular biology that measures the average gene expression profile across a population of cells from a sample, such as tissues, whole organs, or bulk-sorted cell populations [8]. This method provides powerful, large-scale insights into transcriptomes, enabling robust comparisons between different experimental conditions—for instance, healthy versus diseased, or treated versus control groups [8] [7]. Within the context of a broader thesis on bulk RNA sequencing, optimizing the computational pipeline is paramount. A bioinformatics pipeline is a structured sequence of computational processes designed to transform raw sequencing data into interpretable biological findings [78]. The selection of tools within this pipeline is not one-size-fits-all; it must be tailored to the specific biological system and research objectives to ensure accuracy, efficiency, and reproducibility [78]. This guide provides a structured approach to pipeline optimization, detailing tool selection and methodology for diverse research scenarios.
Foundational Principles of Pipeline Optimization
Before selecting specific tools, understanding core optimization principles is crucial for building a robust and efficient bioinformatics pipeline. Optimization extends beyond mere speed; it encompasses accuracy, reproducibility, and resource management [78].
Effective pipeline design rests on several key pillars. Reproducibility is a cornerstone of scientific research, and automated, version-controlled workflows ensure that analyses can be replicated reliably [78]. Scalability ensures the pipeline can handle large datasets from high-throughput sequencing technologies, often requiring parallel computing on high-performance computing (HPC) clusters or cloud platforms [6] [78]. Managing data quality begins with rigorous preprocessing, including quality control, trimming, and filtering, as poor-quality input data inevitably leads to inaccurate results [78]. Finally, proactive resource management—optimizing memory and CPU usage—is essential to prevent computational bottlenecks that can stall analysis [78].
Common challenges in pipeline implementation include batch effects, which are systematic, non-biological variations arising from how samples are collected and processed over time or across multiple sites. A well-designed experiment that randomizes samples and records processing batches can mitigate these effects, and various batch correction software tools exist for in silico correction [13]. Furthermore, the choice between alignment-based and pseudo-alignment quantification methods presents a key trade-off: alignment-based methods (e.g., STAR) generate data useful for extended quality checks, while pseudo-alignment methods (e.g., Salmon) are much faster and can be ideal for analyzing thousands of samples where alignment-based QC metrics are less critical [6].
Tool Selection Strategy for Biological Systems
The optimal bioinformatics pipeline varies significantly depending on the biological context and specific research questions. The table below outlines recommended tools and considerations for different biological systems.
Table 1: Tool Selection Guide for Different Biological Systems and Research Goals
| Biological System / Research Goal | Recommended Alignment/Quantification Tools | Recommended Differential Expression Tools | Key Considerations and Optimizations |
|---|---|---|---|
| Standard Eukaryotic Transcriptomes (e.g., Human, Mouse) | STAR (splice-aware aligner) + Salmon (alignment-based mode) [6] | limma (linear modeling framework) [6] or DESeq2 [60] | The STAR-Salmon hybrid leverages alignment for QC and Salmon's statistical model for count estimation [6]. Requires a genome fasta and GTF annotation file [6]. |
| Gene-Level Differential Expression & Novel Isoform Discovery | STAR for alignment, RSEM for expectation-maximization count estimation [6] | DESeq2 [60] | RSEM models uncertainty in read assignments to transcripts, aiding in isoform-level analysis [6]. |
| Transposable Element (TE) Analysis | STAR (with high multi-mapping limits, e.g., --outFilterMultimapNmax 100), TEcount from TEToolkit for quantification [60] |
DESeq2 [60] | Standard gene annotation (e.g., Gencode) and a dedicated TE GTF file are required [60]. Multi-mapping reads must be accounted for. |
| Large-Scale Drug Screening (Cell Lines) | Salmon in pseudoalignment mode on fastq files [6] | limma or DESeq2 [13] | Pseudoalignment offers significant speed advantages for hundreds/thousands of samples. 3'-Seq library prep (e.g., QuantSeq) from lysates can bypass RNA extraction [13]. |
| Microbiome/Metagenomic Analysis | Specialized tools for taxonomic classification and functional annotation (e.g., MetaPhlAn, HUMAnN) | Tools tailored for metagenomic data | Pipeline can be streamlined by using a single tool for both taxonomic and functional analysis [78]. |
Workflow Management and Integrated Pipelines
For end-to-end analysis, leveraging workflow management systems is a best practice for ensuring reproducibility and portability across computing environments. Nextflow is a prominent workflow language that automates multi-step data analysis, making it easy to adapt pipelines for HPC or cloud environments [6]. Pre-built, community-vetted pipelines like the nf-core/rnaseq workflow provide excellent starting points, automating steps from raw fastq files to count matrices and incorporating tools like STAR and Salmon by default [6]. Similarly, organizations like NASA's GeneLab have made their bulk RNA-Seq consensus processing pipelines, wrapped in Nextflow, publicly available on GitHub, serving as valuable optimized templates [7].
Detailed Experimental Protocols and Methodologies
This section provides detailed protocols for two common and distinct analytical scenarios: a standard differential expression analysis and a specialized transposable element analysis.
Protocol 1: Standard Differential Expression Analysis with nf-core/rnaseq and limma
This protocol is designed for identifying genes differentially expressed between conditions in a standard eukaryotic system (e.g., human cell lines or tissue) [6].
1. Data Preparation and Input:
- Input Data: A set of paired-end RNA-seq fastq files and a sample sheet in nf-core format with columns:
sample,fastq_1,fastq_2, andstrandedness(recommended to set as "auto") [6]. - Reference Files: A genome fasta file and a GTF annotation file for the target species [6].
2. Expression Quantification with nf-core/rnaseq:
- Execute the nf-core/rnaseq workflow with the "STAR-salmon" option. This workflow performs the following steps automatically [6]:
- Spliced Alignment: Aligns reads to the genome using STAR, generating BAM files for quality control.
- Projection and Quantification: Projects genome alignments onto the transcriptome and performs alignment-based quantification with Salmon.
- Output: Produces a gene-level count matrix, where rows correspond to genes and columns to samples.
3. Differential Expression Analysis in R:
- Use the gene-level count matrix as input for the
limmapackage in R [6]. - The linear modeling framework in
limmais used to test the null hypothesis that expression of individual genes does not vary between conditions. - Statistical inference, creation of figures (e.g., volcano plots, MA plots), and interpretation of results are performed in this environment [6].
Protocol 2: Transposable Element (TE) Subfamily Quantification
This protocol is designed for the differential expression analysis of TE subfamilies, which requires specific parameter adjustments to account for repetitive genomic sequences [60].
1. Read Mapping with STAR:
- Map reads using the STAR aligner with an increased multi-mapping limit.
- Critical Parameters:
--outFilterMultimapNmax 100(Allows up to 100 mapping loci per read)--winAnchorMultimapNmax 200(Increases the number of anchors for multi-mapping reads) [60]
2. TE Quantification with TEcount:
- Run
TEcountfrom the TEToolkit inmultimode. - Input Files:
--GTF: A standard gene annotation file (e.g., Gencode).--TE: A provided TE annotation file in GTF format.
- This step generates a read count matrix for TE subfamilies [60].
3. Differential TE Expression with DESeq2:
- Use the TE count matrix as input for DESeq2.
- Perform differential expression analysis similarly to gene-level analysis.
- Normalization: For visualization, TE counts can be normalized using the size factors (sample distances) calculated by DESeq2 from a concurrently run gene-level quantification with unique mappers [60].
Table 2: Research Reagent Solutions for Bulk RNA-Seq Experiments
| Reagent / Material | Function / Application | Considerations |
|---|---|---|
| PolyA Selection Beads | Enriches for messenger RNA (mRNA) by binding the poly-adenylate tail [7]. | Standard for most mRNA-seq; may miss non-polyadenylated RNAs. |
| Ribo-depletion Kits | Removes ribosomal RNA (rRNA) which constitutes >80% of total RNA, enriching for other RNA species [7]. | Preferred for degraded samples (e.g., FFPE) or when studying non-coding RNAs. |
| Spike-in Controls (e.g., SIRVs) | Artificial RNA mixes of known concentration and sequence [13]. | Used to measure assay performance, normalize data, assess technical variability, and act as a quality control for large-scale experiments. |
| Strand-Specific Library Prep Kits | Preserves the original strand orientation of the RNA transcript during cDNA library preparation. | Crucial for determining the direction of transcription and resolving overlapping genes. |
| 3'-Seq Kits (e.g., QuantSeq) | Targets the 3' end of transcripts for library preparation [13]. | Ideal for large-scale drug screens; enables cost-effective, high-throughput expression profiling and can be performed directly from cell lysates. |
Workflow Visualization and Logical Pathways
The following diagrams, generated with Graphviz DOT language, illustrate the logical structure and data flow of the optimized pipelines described in this guide.
Standard Bulk RNA-seq Analysis
Tool Selection Decision Logic
Optimizing a bulk RNA-seq pipeline is an exercise in strategic compromise, balancing computational efficiency, statistical robustness, and biological specificity. There is no universal solution; the optimal toolchain must be selected based on the biological system, the specific research question, and practical constraints like sample size and computational resources [6] [78] [13]. As demonstrated, best practices involve leveraging integrated workflows like nf-core for reproducibility and carefully choosing between alignment and pseudo-alignment methods based on the need for quality metrics versus speed [6].
The future of bioinformatics pipeline optimization is being shaped by emerging technologies. Increased automation and the development of real-time analysis capabilities are on the horizon [78]. Furthermore, the field is exploring the potential of quantum computing to solve core biological modeling problems, such as metabolic flux balance analysis, which could eventually revolutionize how we simulate and analyze complex biological networks at scale [79]. By adhering to the principles and protocols outlined in this guide, researchers can build robust, optimized pipelines that yield reliable biological insights and form a solid foundation for leveraging these future technological advances.
Bulk RNA sequencing (RNA-seq) is an indispensable tool in molecular biology that provides a comprehensive snapshot of gene expression in biological samples, enabling discoveries across biomedical research and drug development [7] [80]. Despite its established position, the analytical journey from raw sequencing data to biologically meaningful results is fraught with technical challenges that can compromise data integrity. Two of the most pervasive issues researchers encounter are low-quality data, stemming from preanalytical to analytical variability, and mapping challenges, where reads are incorrectly assigned to their genomic origins [81] [82]. Within the broader thesis of how bulk RNA sequencing works, this guide addresses these critical bottlenecks, providing a systematic framework for identifying, troubleshooting, and resolving these issues to ensure the generation of robust and reliable transcriptomic data.
Assessing RNA-seq Data Quality
Key Quality Control Metrics
A successful bulk RNA-seq analysis hinges on the initial assessment of data quality through a panel of technical metrics. Research demonstrates that no single metric is sufficient to predict sample quality; instead, an integrated approach is required [81]. The most informative metrics for identifying low-quality samples, as identified by Hamilton et al., include the percentage and absolute number of uniquely aligned reads, the percentage of ribosomal RNA (rRNA) reads, the number of detected genes, and the Area Under the Gene Body Coverage Curve (AUC-GBC) [81].
Table: Key Bulk RNA-seq QC Metrics and Interpretation
| Metric Category | Specific Metric | Interpretation | Optimal Range/Value |
|---|---|---|---|
| Alignment | % Uniquely Aligned Reads | Measures mappability; lower values suggest degradation or contamination. | Ideally >70-80% [81] |
| % rRNA Reads | High levels indicate inefficient rRNA depletion. | Should be low (e.g., <5-10%) [7] | |
| Gene Detection | # Detected Genes | Count of genes with non-zero expression; low counts suggest low quality. | Compare to reference dataset [81] |
| Coverage | Area Under the Gene Body Coverage (AUC-GBC) | Newly developed metric; assesses 5' to 3' coverage uniformity. | Correlates strongly with sample quality [81] |
| Preanalytical | RNA Integrity Number (RIN) | Measures RNA degradation; critical for biobanked samples. | >7 is often recommended [83] |
| Genomic DNA (gDNA) Contamination | Presence of gDNA can lead to spurious alignments. | Should be minimal; may require DNase treatment [83] |
An Integrated QC Framework
For clinical and biomarker discovery studies, implementing a multilayered, end-to-end QC framework across preanalytical, analytical, and postanalytical processes is paramount [83]. Preanalytical factors, including specimen collection, RNA integrity, and genomic DNA contamination, often exhibit the highest failure rates [83]. For instance, the addition of a secondary DNase treatment has been shown to significantly reduce genomic DNA levels, which in turn lowers intergenic read alignment and improves downstream analysis [83].
Advanced approaches now leverage machine learning to predict sample quality by integrating multiple QC metrics. Tools like the Quality Control Diagnostic Renderer (QC-DR) are designed to visualize a comprehensive panel of these metrics and flag samples with aberrant values when compared to a reference dataset [81]. Such models perform robustly even when tested on independent datasets with differing distributions of QC metrics, underscoring the power of a multi-metric, integrated approach over relying on individual thresholds [81].
Addressing Mapping Challenges
A primary challenge in RNA-seq analysis is the accurate alignment of sequencing reads, a process complicated by the complex structure of the transcriptome. A significant issue arises from the coexistence of nascent (unprocessed) and mature (processed) messenger RNA within a sample [82]. Traditional reference transcriptomes are built solely from mature mRNA sequences, which can lead to two major problems:
- Mismapping: Reads originating from nascent mRNA, which includes intronic regions, may be incorrectly forced to map to a mature transcript [82].
- Unmatched Reads: Reads that do not find a perfect match within the defined mature transcriptome are often excluded from analysis, leading to a loss of biologically relevant information [82].
To resolve these issues, a two-pronged strategy has been proposed. First, expanding the transcriptome's "region of interest" to include both nascent and mature mRNA provides a more comprehensive framework for capturing the full diversity of RNA molecules [82]. Second, the use of distinguishing flanking k-mers (DFKs)—short, unique nucleotide sequences that can differentiate between nascent and mature RNA—serves as a "background filter" to enhance mapping accuracy and resolve ambiguous reads [82].
Another best practice involves a hybrid quantification approach. This method first uses a splice-aware aligner like STAR to map reads to the genome, generating alignment files (BAM) crucial for detailed quality control [6]. These genomic alignments are then projected onto the transcriptome and fed into alignment-based quantification tools like Salmon, which use statistical models to handle the uncertainty of assigning reads to transcripts, thereby producing more accurate expression estimates [6].
Recommended Computational Workflows
Automated bioinformatics workflows streamline the data preparation process, ensuring reproducibility and robustness. The nf-core RNA-seq pipeline is a widely adopted Nextflow workflow that implements best practices [6]. Its "STAR-salmon" option is particularly recommended as it executes a series of optimized steps: it performs spliced alignment with STAR, projects the genomic alignments to the transcriptome, and then runs alignment-based quantification with Salmon [6]. This integrated process efficiently generates both the crucial QC metrics from the BAM files and a statistically robust gene-level count matrix ready for differential expression analysis.
The Scientist's Toolkit
Successful bulk RNA-seq experiments rely on a combination of robust computational tools, high-quality reagents, and well-validated protocols. The table below details key resources for troubleshooting low-quality data and mapping challenges.
Table: Essential Research Reagent and Computational Solutions
| Category | Item | Specific Example / Tool | Function / Application |
|---|---|---|---|
| Wet-Lab Reagents | RNA Stabilization Tubes | PAXgene Blood RNA Tubes | Preserves RNA integrity in whole blood samples [83] |
| DNase Treatment Kit | Secondary DNase Treatment | Reduces genomic DNA contamination, lowering intergenic reads [83] | |
| rRNA Depletion / PolyA Selection Kits | Various commercial kits | Enriches for mRNA, reducing %rRNA reads [7] | |
| Computational Tools | Quality Control & Visualization | QC-DR (Quality Control Diagnostic Renderer) | Integrates and visualizes multiple QC metrics to flag outliers [81] |
| dittoSeq | Universal, user-friendly R toolkit for visualizing QC and analysis results [84] [85] | ||
| Read Alignment | STAR | Splice-aware aligner for mapping RNA-seq reads to a reference genome [6] | |
| Expression Quantification | Salmon | Performs fast, accurate alignment-based or pseudoalignment-based quantification [6] | |
| Automated Workflow | nf-core/rnaseq (STAR-salmon) | End-to-end automated pipeline for reproducible RNA-seq data processing [6] | |
| Experimental Protocols | End-to-End QC Framework | Multilayered QC (Preanalytical to Postanalytical) | Systematic framework for enhancing confidence in RNA-seq biomarker discovery [83] |
Navigating the challenges of low-quality data and mapping errors is fundamental to unlocking the full potential of bulk RNA-seq. As we have detailed, a successful strategy moves beyond relying on single metrics and adopts an integrated, multi-layered approach. This involves leveraging machine learning-powered tools like QC-DR for robust quality assessment [81], implementing sophisticated mapping strategies that account for transcriptional complexity [82], and utilizing automated, best-practice workflows like nf-core/rnaseq for reproducible analysis [6]. For the field to advance, particularly in clinical translation, the development and adoption of standardized end-to-end QC frameworks, as described in clinical validation studies [83], will be crucial. By adhering to these rigorous methodological practices, researchers can mitigate technical variability, enhance the reliability of their gene expression data, and ensure their findings are built upon a solid analytical foundation.
Validating and Contextualizing Bulk RNA-Seq Findings
Bulk RNA sequencing (bulk RNA-seq) provides a population-average view of gene expression across all cells in a complex biological sample. While this technology delivers powerful insights into transcriptomic profiles, the averaging effect inherently masks cellular heterogeneity and can obscure biologically significant signals originating from specific cell subpopulations. This limitation makes robust technical validation not merely beneficial but essential for confirming key findings and ensuring research conclusions are reliable, reproducible, and biologically meaningful. Within the broader thesis of how bulk RNA sequencing works in research, validation serves as the critical bridge between raw genomic data and scientifically sound interpretation, particularly in translational applications like drug discovery where decisions have significant clinical and financial implications [13] [8].
This guide details a multi-faceted framework for validating bulk RNA-seq findings, encompassing orthogonal assay confirmation, advanced computational deconvolution, and best-practice experimental design to maximize the reliability of generated data.
A Framework for Analytical Validation
A comprehensive, multi-step validation strategy is fundamental for establishing confidence in bulk RNA-seq results. A robust framework should integrate analytical validation using reference standards, orthogonal confirmation with independent methodologies, and finally, assessment of clinical or biological utility in real-world scenarios [86].
Table 1: Pillars of a Bulk RNA-seq Validation Strategy
| Validation Pillar | Description | Key Tools/Methods | Primary Objective |
|---|---|---|---|
| Analytical Validation | Assess assay performance using benchmarks and reference standards. | Cell lines at varying purities; synthetic spike-in controls (e.g., SIRVs) [86] [13]. | Determine accuracy, sensitivity, and specificity of the sequencing assay itself. |
| Orthogonal Confirmation | Verify key findings using a different technological principle. | qRT-PCR, droplet digital PCR (ddPCR), single-cell RNA-seq (scRNA-seq) [86] [8]. | Provide independent, technical confirmation of specific gene expression changes or variants. |
| Clinical/Biological Utility | Evaluate the real-world impact and applicability of the findings. | Correlation with patient outcomes; functional experiments (e.g., knock-down, knock-out) [86] [87]. | Establish the biological relevance and potential translational value of the results. |
Validation of Specific Data Types
Bulk RNA-seq delivers diverse data types, each requiring tailored validation approaches to confirm its integrity and biological significance.
Gene Expression and Differential Expression
Validation of gene-level findings is the most common practice. Differential expression analysis, typically performed with tools like limma, should be confirmed by:
- Orthogonal Quantification: Using qRT-PCR or ddPCR for a subset of differentially expressed genes (DEGs). This is considered the gold standard for technical confirmation [6].
- Spike-In Controls: Employing artificial RNA spike-in controls (e.g., SIRVs) during library preparation. These act as an internal standard to measure assay performance, control for technical variability, and aid in normalization [13].
- Biological Replication: Ensuring an adequate number of biological replicates (typically 3-8 per condition) during experimental design is a foundational form of validation, as it ensures findings are consistent and generalizable beyond individual samples [13].
Genetic Variants and Fusion Transcripts
Identifying sequence variants and gene fusions from bulk RNA-seq requires specialized analytical approaches and stringent validation.
- Orthogonal DNA Sequencing: Confirming RNA-derived somatic single nucleotide variants (SNVs) and insertions/deletions (INDELs) with DNA-level assays, such as whole exome sequencing (WES), provides a higher level of certainty [86].
- Integrated DNA-RNA Assays: Using validated assays that combine RNA-seq and WES from a single sample improves the detection of clinically relevant alterations and can recover variants missed by DNA-only approaches. These integrated workflows facilitate direct correlation of somatic alterations with gene expression changes [86].
- Dedicated Variant Calling Pipelines: Utilizing gold-standard workflows like the GATK best practices for RNA-seq variant calling ensures high-quality variant identification. Tools like
RnaXtractautomate this process within a reproducible Snakemake framework, integrating quality control, variant calling, and filtration specific to SNPs and INDELs [88].
Cellular Heterogeneity via Computational Deconvolution
A significant advancement in bulk RNA-seq analysis is the computational estimation of cell type composition and cell type-specific (CTS) expression, a process known as deconvolution.
- Single-Cell RNA-seq as a Reference: Deconvolution methods leverage scRNA-seq data from a similar tissue as a reference to infer the proportions and, in some cases, the expression profiles of specific cell types within a bulk sample [89] [88].
- Validation with scRNA-seq: The most direct way to validate deconvolution results is by comparing them with true cellular proportions or CTS expression profiles obtained from scRNA-seq analysis of matched or similar samples [89] [8].
- Advanced Deconvolution Tools: State-of-the-art methods like
EPIC-unmixuse a two-step empirical Bayesian framework to integrate single-cell and bulk data, accounting for differences between reference and target datasets. This approach has been shown to outperform other methods in accuracy for inferring CTS expression [89]. Other powerful tools includeCIBERSORTxandEcoTyper, which can be integrated into automated pipelines likeRnaXtractto decode cellular heterogeneity from bulk data [88].
Detailed Experimental Protocols
Protocol: Deconvolution of Bulk RNA-seq Data using a Single-Cell Reference
This protocol outlines the steps to infer cell type composition and cell type-specific expression from bulk RNA-seq data.
Step 1: Obtain a Single-Cell Reference Dataset
- Acquire a scRNA-seq or snRNA-seq dataset from a biologically similar tissue. Public repositories like the Gene Expression Omnibus (GEO) are common sources [89] [87].
- Perform standard quality control and cell type annotation on the reference data using tools like the
Seuratworkflow to define major cell populations [87].
Step 2: Preprocess Bulk RNA-seq Data
Step 3: Apply a Deconvolution Algorithm
- Use a method like
EPIC-unmix,CIBERSORTx, orbMIND. The following is an example usingEPIC-unmix: - Inputs: The normalized bulk expression matrix and cell type fractions for each sample. (Cell type fractions can be estimated from the bulk data using a tool like
MuSiCand the single-cell reference) [89]. - Execution: Run the two-step empirical Bayesian inference. The first step uses the single-cell reference to build a prior for CTS expression. The second step refines this for the target bulk data, making it adaptive to dataset-specific differences [89].
- Gene Selection: For optimal performance, apply a gene selection strategy focused on known cell-type marker genes, as this significantly improves deconvolution accuracy [89].
- Use a method like
Step 4: Validate and Interpret Results
- Compare the estimated cell type proportions with known biology or histopathological data if available.
- For CTS expression, validate key findings using external datasets or functional experiments. The output will be k sample-by-gene matrices of expression values for k cell types [89].
Protocol: Orthogonal Validation of Differential Expression by qRT-PCR
Step 1: Select Target Genes
- Choose a panel of significant DEGs from the bulk RNA-seq analysis, including both up- and down-regulated genes.
Step 2: cDNA Synthesis
- Using the same original RNA samples that were sequenced, synthesize cDNA with a reverse transcription kit.
Step 3: qRT-PCR Assay
- Design and validate primer pairs for each target gene and for stable reference genes (e.g., GAPDH, ACTB).
- Run qRT-PCR reactions in technical triplicates for each biological sample.
- Calculate relative gene expression using the ΔΔCt method.
Step 4: Correlation Analysis
- Compare the log2 fold-changes obtained from the bulk RNA-seq analysis with the log2 fold-changes from the qRT-PCR data. A strong positive correlation validates the sequencing results.
Visualizing Workflows and Signaling Pathways
Bulk RNA-seq Validation Workflow
The following diagram illustrates the logical relationships and pathways for a comprehensive bulk RNA-seq validation strategy.
Integrated DNA-RNA Sequencing Assay Workflow
This diagram details the workflow for a validated integrated sequencing assay that combines DNA and RNA data from a single tumor sample.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Validation
| Category | Item | Function in Validation |
|---|---|---|
| Reference Standards | Cell lines at varying purities [86] | Analytical validation controls for sensitivity and specificity. |
| Synthetic spike-in RNA (e.g., SIRVs) [13] | Internal controls for normalization and technical performance monitoring. | |
| Orthogonal Assay Kits | qRT-PCR or ddPCR reagents [6] | Independent, highly sensitive confirmation of gene expression levels. |
| Library Prep Kits | Stranded mRNA kits (e.g., Illumina TruSeq) [86] | Ensure high-quality, strand-specific RNA-seq libraries. |
| Extraction-free 3'-Seq kits (e.g., QuantSeq) [13] | Streamlined library prep for large-scale studies like drug screens. | |
| Bioinformatics Tools | Snakemake workflow manager [88] | Ensures reproducible and automated analysis pipelines. |
| CIBERSORTx / EcoTyper [89] [88] | Enables cell-type deconvolution from bulk RNA-seq data. | |
| GATK variant calling toolkit [88] | Gold-standard for identifying genetic variants from RNA-seq data. | |
| RnaXtract pipeline [88] | All-in-one tool for expression, variant, and deconvolution analysis. |
Technical validation is the cornerstone of robust and interpretable bulk RNA-seq research. By systematically implementing a framework that combines analytical rigor, orthogonal confirmation, and advanced computational methods like deconvolution, researchers can transcend the limitations of population-averaged data. This approach unlocks deeper, more reliable biological insights, confidently revealing the complex cellular narratives hidden within bulk tissue samples. As the field progresses, integrated workflows and sophisticated validation strategies will be paramount in translating bulk RNA-seq findings into meaningful advancements in basic science and drug development.
Next-generation sequencing technologies have revolutionized the field of transcriptomics, with bulk RNA sequencing (bulk RNA-Seq) and single-cell RNA sequencing (scRNA-Seq) emerging as pivotal methodologies for gene expression analysis. While both techniques share the common goal of transcriptome profiling, they differ fundamentally in resolution, applications, and technical considerations. Bulk RNA-Seq provides a population-average view of gene expression from a tissue or cell population, whereas scRNA-Seq enables the investigation of transcriptional profiles at individual cell resolution. This technical guide provides an in-depth comparative framework for these two powerful approaches, focusing on their experimental paradigms, applications in research and drug discovery, and methodological considerations to inform selection for specific research objectives.
Fundamental Technical Differences
The core distinction between bulk and single-cell RNA sequencing lies in their resolution and sample processing approaches. Bulk RNA-Seq analyzes the averaged gene expression from a population of cells, typically thousands to millions, processed collectively as a single sample. The RNA from all cells is extracted and pooled together before library preparation and sequencing, resulting in a composite expression profile that represents the entire cell population. This method is particularly effective for homogeneous samples or when studying overall transcriptional changes between different conditions.
In contrast, single-cell RNA-Seq isolates individual cells before RNA capture and sequencing. Each cell's transcriptome is processed separately, often utilizing cell barcoding strategies to track the cellular origin of each transcript. The 10X Genomics Chromium system, for instance, employs microfluidic chips to partition single cells into gel bead-in-emulsions (GEMs), where each gel bead contains a cell-specific barcode that labels all transcripts from that individual cell. This approach preserves cellular identity throughout the sequencing workflow, enabling the resolution of cellular heterogeneity within complex tissues.
The methodological divergence creates significant implications for data interpretation. Bulk RNA-Seq provides higher gene detection sensitivity per sample, with one study reporting median detection of 13,378 genes compared to 3,361 genes in matched scRNA-Seq samples. However, scRNA-Seq excels in detecting cellular subpopulations and rare cell types that are masked in bulk sequencing due to their low abundance.
Comparative Analysis: Key Parameters
Table 1: Technical Comparison of Bulk RNA-Seq vs. Single-Cell RNA-Seq
| Parameter | Bulk RNA-Seq | Single-Cell RNA-Seq |
|---|---|---|
| Resolution | Population average (multiple cells) | Individual cell level |
| Cost per Sample | Lower (~$300/sample) | Higher (~$500-$2000/sample) |
| Data Complexity | Lower, simpler analysis | Higher, requires specialized computational methods |
| Cell Heterogeneity Detection | Limited, masks diversity | High, reveals cellular subpopulations |
| Sample Input Requirement | Higher (micrograms of RNA) | Lower (single cell or picograms of RNA) |
| Rare Cell Type Detection | Limited, masked by abundant cells | Possible, can identify rare populations |
| Gene Detection Sensitivity | Higher (detects more genes per sample) | Lower due to dropout events and sparsity |
| Splicing Analysis | More comprehensive | Limited |
| Typical Applications | Differential expression, biomarker discovery, transcriptome annotation | Cell typing, developmental trajectories, tumor heterogeneity, immune profiling |
Table 2: Applications in Drug Discovery and Development
| Application | Bulk RNA-Seq Utility | Single-Cell RNA-Seq Utility |
|---|---|---|
| Target Identification | Gene expression profiling between conditions | Cell-type-specific target discovery in complex tissues |
| Biomarker Discovery | Population-level biomarkers | Cell-type-specific biomarkers, rare cell population markers |
| Tumor Microenvironment | Overall expression signatures | Dissecting cellular heterogeneity, stromal-immune interactions |
| Drug Mechanism Studies | Average transcriptomic responses | Heterogeneous drug responses across cell types |
| Pharmacogenomics | Population-level expression quantitative trait loci (eQTLs) | Cell-type-specific eQTL mapping |
| Toxicity Assessment | Overall pathway perturbations | Identifying sensitive cell populations |
Experimental Workflows and Protocols
Bulk RNA-Seq Experimental Workflow
The standard bulk RNA-Seq protocol begins with sample collection from tissues or cell cultures, followed by total RNA extraction. RNA quality assessment is critical, typically measured by RNA Integrity Number (RIN), with values above 6 considered suitable for sequencing. Library preparation involves several key steps: (1) RNA fragmentation through enzymatic, chemical, or physical methods; (2) cDNA synthesis via reverse transcription; (3) adapter ligation for sequencing compatibility; and (4) PCR amplification to generate sufficient material for sequencing. rRNA depletion or mRNA enrichment using poly(A) selection is commonly performed to focus on protein-coding transcripts. The final library is sequenced using short-read (Illumina) or long-read (PacBio, Oxford Nanopore) platforms, with paired-end sequencing preferred for better transcript assembly and isoform identification.
Single-Cell RNA-Seq Experimental Workflow
The scRNA-Seq workflow introduces critical steps for cellular resolution. Sample dissociation into viable single-cell suspensions is the first crucial step, requiring optimization to maintain cell viability while avoiding transcriptional stress responses. The 10X Genomics Chromium system utilizes microfluidic partitioning where single cells are encapsulated in oil-water emulsion droplets (GEMs) together with barcoded gel beads. Within each GEM, cells are lysed, and mRNA transcripts are tagged with cell barcodes and unique molecular identifiers (UMIs) during reverse transcription. After breaking emulsions, barcoded cDNA is amplified and processed into sequencing libraries. The resulting data undergoes specialized computational processing including cell calling, UMI counting, quality control to remove doublets and empty droplets, normalization, and dimensionality reduction for visualization and clustering.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for RNA Sequencing
| Reagent/Platform | Function | Examples/Providers |
|---|---|---|
| Cell Isolation Kits | Tissue dissociation into single cells for scRNA-Seq | Enzymatic (collagenase, trypsin) or mechanical dissociation kits |
| Viability Stains | Distinguish live/dead cells during quality control | Trypan blue, propidium iodide, fluorescent viability dyes |
| Barcoded Gel Beads | Single-cell partitioning and mRNA barcoding | 10X Genomics Barcoded Gel Beads, Parse Biosciences Evercode combinatorial barcodes |
| Library Prep Kits | Convert RNA to sequencing-ready libraries | Illumina TruSeq, 10X Genomics Single Cell 3' / 5' Kits, SMART-Seq2 reagents |
| mRNA Enrichment | Select for polyadenylated transcripts | Poly(A) selection beads, oligo(dT) primers |
| rRNA Depletion Kits | Remove ribosomal RNA to enhance signal | Ribozero, NEBNext rRNA Depletion Kit |
| UMI Reagents | Unique Molecular Identifiers for quantitative accuracy | Custom UMI oligos, commercial UMI kits |
| Platform Instruments | Single-cell partitioning and library preparation | 10X Genomics Chromium Controller/Connect, Chromium X series, Parse Biosciences platforms |
Applications in Research and Drug Discovery
Bulk RNA-Seq Applications
Bulk RNA-Seq remains the workhorse for numerous transcriptomic applications where population-level insights are sufficient or preferred. In differential gene expression analysis, it robustly identifies transcriptional changes between conditions, such as disease versus healthy states or treated versus control samples. For biomarker discovery, bulk approaches have successfully identified prognostic gene signatures across cancer types, though with limitations in reproducibility due to tumor heterogeneity. Bulk RNA-Seq excels in gene fusion detection, with studies of nearly 7,000 cancer samples from The Cancer Genome Atlas revealing novel kinase fusions with therapeutic implications. Additionally, it provides comprehensive splicing analysis and isoform characterization, offering insights into transcript diversity that can be challenging with standard scRNA-Seq protocols.
Single-Cell RNA-Seq Applications
Single-cell technologies have transformed our ability to dissect cellular heterogeneity in complex biological systems. In oncology, scRNA-Seq has revealed intratumoral heterogeneity in glioblastoma and identified rare drug-resistant subpopulations in melanoma that were masked in bulk analyses. For immunology, researchers have discovered previously unknown dendritic cell and monocyte subsets in human blood, revolutionizing our understanding of immune cell diversity. In developmental biology, scRNA-Seq enables the reconstruction of differentiation trajectories and lineage relationships. The technology has been particularly impactful in neurology, where it has characterized diverse neuronal and glial cell types in the brain. In drug discovery, scRNA-Seq helps identify cell-type-specific drug targets and understand heterogeneous therapeutic responses, with one study identifying CFTR-expressing pulmonary ionocytes (1 in 200 cells) as potential mediators of cystic fibrosis pathology.
Integrated Approaches in Precision Medicine
Increasingly, researchers are leveraging both technologies in complementary approaches. The scDEAL framework demonstrates how transfer learning can integrate large-scale bulk cell line drug response data with scRNA-Seq to predict single-cell drug sensitivity. This integration addresses the limitation of insufficient training data for scRNA-Seq drug response prediction by transferring knowledge from bulk repositories like GDSC and CCLE. Such hybrid approaches exemplify the powerful synergy between these technologies, combining the statistical power of bulk sequencing with the resolution of single-cell methods.
Selection Guidelines and Future Perspectives
The choice between bulk and single-cell RNA-Seq depends on research questions, budget, and sample characteristics. Bulk RNA-Seq is recommended when: studying homogeneous cell populations, conducting large-scale cohort studies with budget constraints, focusing on differential expression without cellular heterogeneity concerns, requiring high sensitivity for low-abundance transcripts, or performing comprehensive splicing and isoform analysis. Single-cell RNA-Seq is preferable when: investigating heterogeneous tissues (tumors, immune cells, nervous system), identifying rare cell populations or transient states, reconstructing developmental trajectories, studying cell-type-specific responses to perturbations, or analyzing samples with limited cellular material.
Future methodological developments focus on overcoming current limitations. Multi-omic integrations combine scRNA-Seq with epigenetic (scATAC-Seq) and proteomic (CITE-Seq) profiling for comprehensive cellular characterization. Spatial transcriptomics technologies address the loss of spatial context in scRNA-Seq by mapping gene expression within tissue architecture. Computational advances continue to improve data imputation, integration, and interpretation for both bulk and single-cell approaches. As sequencing costs decrease and methodologies mature, the complementary application of these powerful technologies will further accelerate discoveries in basic research and therapeutic development.
Bulk RNA sequencing (Bulk RNA-seq) is a foundational genomic technique for transcriptomic analysis that measures the average expression level of individual genes across hundreds to millions of input cells from pooled cell populations, tissue sections, or biopsies [24]. The "resolution" in this context refers to the technical and analytical sensitivity of the experiment to detect true biological signals, such as differentially expressed genes (DEGs), against a background of technical noise and biological variability. Understanding the trade-offs inherent to this resolution is critical for researchers, scientists, and drug development professionals who must design experiments that are both powerful and cost-effective, ensuring accurate biological interpretations.
This technical guide frames these trade-offs within a broader thesis on how bulk RNA sequencing works, dissecting the core experimental and analytical steps where decisions directly impact the resolution and reliability of the final results. Proper experimental design and a deep understanding of the following trade-offs are essential to avoid erroneous conclusions and to maximize the return on often substantial research investments.
Key Experimental Protocols in Bulk RNA-Seq
A standardized protocol is crucial for generating high-quality, reproducible data. The following outlines a typical bulk RNA-seq workflow, highlighting steps where methodological choices influence experimental outcomes [10] [24].
Sample Preparation and Library Construction
The process begins with extracting total RNA from the biological sample. The quality of the starting material is paramount; it is typically assessed using a Bioanalyzer to generate an RNA Integrity Number (RIN). A RIN score of 7 or higher is generally considered sufficient for library construction [90].
Key steps include:
- mRNA Enrichment: mRNA is isolated from total RNA using oligo-dT primers to capture polyadenylated transcripts. Alternatively, ribosomal RNA (rRNA) depletion is used for non-coding RNA or total RNA-seq workflows [10] [90].
- cDNA Synthesis: The enriched mRNA is reverse-transcribed into complementary DNA (cDNA), followed by second-strand synthesis to generate double-stranded cDNA [24].
- Library Preparation and Barcoding: The cDNA fragments are ligated with adapters, which often include unique molecular barcodes. This allows multiple samples (libraries) to be pooled together in a single sequencing lane, a process known as multiplexing, which reduces costs and controls for batch effects [24]. The final sequenceable library is then quality-controlled for fragment size and concentration, typically using an Agilent TapeStation system [24].
Sequencing and Primary Data Analysis
The pooled libraries are sequenced using high-throughput platforms, most commonly Next-Generation Sequencing (NGS) technologies like Illumina [91]. The primary data analysis involves:
- Demultiplexing: Raw sequencing data (in BCL format) is converted to FASTQ files, and reads are assigned to their original samples based on their unique barcodes [10].
- Alignment and Quantification: The sequenced reads in the FASTQ files are aligned to a reference genome (e.g., mm10 for mouse) using aligners like STAR. Following alignment, tools like HTSeq-count are used to generate a count matrix, which tabulates the number of reads uniquely assigned to each gene for every sample [10]. This count matrix is the fundamental input for downstream differential expression analysis.
Critical Resolution Trade-offs in Bulk RNA-Seq
The design and execution of a bulk RNA-seq experiment involve navigating several key trade-offs that directly impact the effective resolution and interpretability of the data.
Trade-off 1: Sequencing Depth vs. Cost and Multiplexing
Sequencing depth refers to the number of reads sequenced per sample. Higher depth increases the statistical power to detect lowly expressed genes and improves the quantification accuracy for all genes. However, this comes with a direct and substantial increase in cost. Furthermore, devoting more reads to one sample within a multiplexed run reduces the number of samples that can be processed simultaneously. The goal is to achieve sufficient depth to answer the biological question without wasteful oversequencing. For standard differential expression analyses, 20-50 million reads per sample is often adequate, though this depends on the complexity of the transcriptome and the expected effect sizes [90].
Trade-off 2: Statistical Sensitivity vs. Specificity in Differential Expression
A core analytical challenge is balancing Type I (false positives) and Type II (false negatives) errors. This is managed through statistical thresholds during differential expression testing, which is commonly performed with tools like DESeq2 [19].
- P-value and Multiple Testing Correction: While a p-value cutoff of 0.05 is standard for a single test, an RNA-seq experiment involves thousands of simultaneous tests (one per gene). Without correction, this leads to a proliferation of false positives. To control this, a False Discovery Rate (FDR) correction is applied, resulting in adjusted p-values (q-values). Using an FDR-adjusted p-value of < 0.05 is the standard benchmark for significance [19] [90]. Although this reduces the number of false positives, it simultaneously increases the risk of false negatives (Type II errors), as some truly differentially expressed genes with weaker signals may not meet this stringent threshold.
- Fold-Change Thresholding: Applying a minimum log2 fold-change threshold (e.g., |log2FC| > 1) alongside significance filters can further improve the biological relevance of results but may also miss genes with subtle but important expression changes.
Table 1: Key Statistical Outputs from a Typical DESeq2 Analysis and Their Interpretation
| Output Column | Description | Role in Resolution Trade-off |
|---|---|---|
| baseMean | The mean normalized expression of the gene across all samples. | Lowly expressed genes have less power to detect differences. |
| log2FoldChange (LFC) | The log2-transformed fold change of expression between groups. | A large magnitude indicates a strong effect size. |
| LFC_shrink | A shrunken LFC value using empirical Bayes methods (e.g., apeglm). | Reduces noise in LFC estimates for low-count genes, improving specificity [19]. |
| pvalue | The raw p-value from the Wald test or similar. | Prone to false positives when thousands of genes are tested. |
| padj | The FDR-adjusted p-value. | Primary filter for significance; a higher stringency reduces false positives at the cost of potential false negatives [19] [90]. |
| svalue | A measure of confidence in the sign (direction) of the LFC. | Provides an alternative, potentially more robust, measure of confidence [19]. |
Trade-off 3: Sample Replication vs. Resource Constraints
Biological replication (using multiple independent biological samples per group) is non-negotiable for drawing statistically sound and generalizable conclusions. It allows for the estimation of biological variance, which is crucial for accurate statistical modeling in tools like DESeq2. While increasing sequencing depth improves the detection of low-abundance transcripts, increasing the number of biological replicates provides greater power to detect smaller, more subtle expression changes that are consistent across a population. Under fixed budgets, a well-powered experiment often prioritizes a greater number of replicates over extreme sequencing depth per sample.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of a bulk RNA-seq experiment relies on a suite of specialized reagents and computational tools.
Table 2: Key Research Reagent Solutions for Bulk RNA-Seq
| Item | Function / Explanation |
|---|---|
| Total RNA | The starting material, ideally with high purity and integrity (RIN > 7) [90]. |
| Oligo-dT Primers / rRNA Depletion Probes | For enriching messenger RNA (mRNA) by binding poly-A tails, or for removing abundant ribosomal RNA to study other RNA species [10] [90]. |
| Reverse Transcriptase | Enzyme for synthesizing complementary DNA (cDNA) from the RNA template [24]. |
| Library Preparation Kit | A commercial kit (e.g., NEBNext) containing enzymes and buffers for cDNA fragmentation, adapter ligation, and PCR amplification [10]. |
| Unique Dual Indexes | Short, unique DNA barcodes added to each sample's cDNA, enabling multiplexing and sample identification after sequencing [24]. |
| Alignment Software (STAR) | Software used to accurately map sequencing reads to a reference genome [10]. |
| Differential Expression Tool (DESeq2) | A statistical software package in R that models count data using a negative binomial distribution to identify differentially expressed genes [19]. |
Workflow and Relationship Visualizations
The following diagrams map the core workflow of a bulk RNA-seq experiment and conceptualize the critical trade-offs discussed.
Bulk RNA-Seq Experimental and Analytical Workflow
Conceptual Framework of Key Trade-offs
Bulk RNA-seq remains a powerful and widely used method for profiling gene expression. Its effective application, however, requires a nuanced understanding of its inherent resolution trade-offs. Researchers must strategically balance sequencing depth against sample replication, and statistical sensitivity against specificity, all within the constraints of a defined budget. There is no universal "best" setting; the optimal design is inherently determined by the specific biological question. By carefully considering these trade-offs during the experimental design and analysis phases, scientists and drug developers can ensure that their bulk RNA-seq data is both robust and interpretable, thereby maximizing the potential for meaningful biological discovery and therapeutic advancement.
Bulk RNA sequencing (RNA-Seq) is a foundational molecular biology technique that measures gene expression across an entire population of cells within a sample, providing powerful, large-scale insights into transcriptional activity [7]. The process involves converting RNA molecules into complementary DNA (cDNA) and sequencing them using next-generation sequencing platforms, typically after removing ribosomal RNA (rRNA) which constitutes over 80% of total RNA [7]. While this method offers broad, quantitative profiling of gene expression, it averages signals across many cells, meaning it cannot capture single-cell heterogeneity [7]. Within systems biology, bulk RNA-Seq serves as a critical component in multi-omics approaches, where its transcriptomic data can be integrated with other molecular data types to construct comprehensive models of biological systems. This integration enables researchers to uncover complex relationships between different layers of molecular regulation, from genetics to proteomics, ultimately advancing our understanding of disease mechanisms and therapeutic development.
Foundational Principles of Bulk RNA-Seq Methodology
Core Workflow and Experimental Design
The bulk RNA-Seq workflow begins with careful experimental design, which is crucial for generating biologically meaningful results. Key considerations include the number and type of replicates, avoidance of confounding factors, and management of batch effects [27]. Biological replicates—different biological samples of the same condition—are absolutely essential as they enable measurement of biological variation between samples [27]. Technical replicates, which use the same biological sample to repeat technical steps, are generally unnecessary with modern RNA-Seq technologies as technical variation is much lower than biological variation [27].
Best practices recommend at least 3 replicates as an absolute minimum, with 4 being the optimum minimum for robust statistical analysis [67]. The relationship between replicates and sequencing depth demonstrates that increasing replicates typically returns more differentially expressed genes than increasing sequencing depth, though higher depth is required for detecting lowly expressed genes or performing isoform-level analysis [27]. For general gene-level differential expression, 15 million reads per sample is often sufficient when there are a good number of replicates (>3), with the ENCODE guidelines suggesting 30 million single-end reads per sample for comprehensive analysis [27].
Critical Experimental Considerations
Confounding occurs when separate effects of two different sources of variation cannot be distinguished in the data [27]. For example, if all control mice were female and all treatment mice were male, the treatment effect would be confounded by sex, making it impossible to differentiate their individual effects [27]. To avoid confounding, researchers should ensure that subjects in each condition are matched for characteristics like sex, age, litter, and batch whenever possible [27].
Batch effects represent another significant challenge in RNA-Seq analyses [27]. These occur when samples are processed at different times, by different people, or with different reagents [27]. The effect of batches on gene expression can often be larger than the effect from the experimental variable of interest [27]. Best practices recommend designing experiments to avoid batches when possible, but if unavoidable, researchers should split replicates of different sample groups across batches and include batch information in experimental metadata so this variation can be accounted for during analysis [27].
Computational Processing and Analysis of Bulk RNA-Seq Data
Standardized Processing Workflows
Processing bulk RNA-Seq data involves several standardized computational steps. The GeneLab consortium has developed a consensus processing pipeline for identifying differentially expressed genes from bulk RNA-Seq data [7]. This workflow begins with quality checking of sample sequences using FastQC for completeness, depth, and read quality, followed by trimming of adapter contamination using Trimmomatic [19]. Sequences are then aligned to the appropriate reference genome using the STAR aligner, and gene quantification is performed using HTSeq-count [19]. For specialized analyses focusing on long noncoding RNAs (lncRNAs), annotations from GENCODE are incorporated during alignment and gene quantification steps [19].
Table 1: Essential Components for Bulk RNA-Seq Analysis
| Component | Description | Function in Analysis |
|---|---|---|
| Raw Data Files (FASTQ) | Unprocessed sequence files containing read data and quality scores | Primary input for alignment and quantification processes |
| Metadata Spreadsheet | Sample information including FASTQ IDs, laboratory IDs, group assignments, and covariates | Links experimental design to raw data; essential for proper statistical modeling |
| Reference Genome | Organism-specific genomic sequence and annotation files | Provides template for read alignment and gene mapping |
| Alignment Software (STAR) | Spliced Transcripts Alignment to a Reference algorithm | Maps sequencing reads to reference genome, accounting for splice junctions |
| Gene Quantification Tool (HTSeq-count) | Python package for counting aligned reads per gene | Generates count matrix for differential expression analysis |
Differential Expression Analysis
For differential gene expression analysis, the DESeq2 package implements a comprehensive statistical framework for analyzing count data from RNA-seq experiments [19]. DESeq2 requires count data as a matrix of integer values where each row represents a gene and each column contains the number of uniquely assigned reads for a sample [19]. The method assumes counts follow a negative binomial distribution and computes normalized counts by scaling with factors that account for differences in sequencing depth between samples [19].
The statistical testing in DESeq2 typically uses the Wald Test, which evaluates the precision of log fold change values to test the hypothesis that a gene is differentially expressed between groups [19]. To address the multiple comparisons problem inherent in testing thousands of genes simultaneously, DESeq2 implements the Benjamini-Hochberg False Discovery Rate (FDR) correction by default, which controls the expected ratio of false positives among significant findings [19]. For more conservative control, Family-wise Error Rate (FWER) corrections can be applied upon request [19].
Effect size estimation is refined using empirical Bayes shrinkage estimators from the apeglm package, which helps prevent extremely large differences that may appear due to technical artifacts rather than biological reality [19]. These methods also compute s-values that provide confidence levels in the direction of log base 2 fold-change values [19].
Automated Analysis and Visualization Tools
Searchlight represents an advanced tool for automating the exploration, visualization, and interpretation (EVI) of bulk RNA-seq data after processing [92]. This freely available pipeline provides comprehensive statistical and visual analysis at global, pathway, and single gene levels through three specialized workflows [92]:
- Normalized Expression (NE) Workflow: Focuses on quality control and experimental overview through expression distribution analysis, principal component analysis (PCA), distance analysis, and highly expressed gene analysis [92].
- Differential Expression (DE) Workflow: Visualizes single differential expression comparisons through MA plots, volcano plots, significant gene heatmaps, spatial analysis by chromosome, and pathway analysis including over-representation analysis (ORA) and upstream regulator analysis (URA) [92].
- Multiple Differential Expression (MDE) Workflow: Explores relationships between two or more sets of differential comparisons through significant gene counts, overlap analysis, fold-change comparisons, and differential expression signature analysis [92].
Searchlight generates R scripts for each plot, allowing researchers to modify and regenerate visualizations, and produces comprehensive HTML reports with interpretation guides [92].
Multi-Omics Integration Approaches
Cross-Modal Data Integration Frameworks
Advanced computational methods enable the integration of bulk RNA-seq data with other omics modalities to uncover novel biological insights. DeepTEX represents one such approach—a multi-omics deep learning method that integrates cross-modal data to investigate T-cell exhaustion heterogeneity in colorectal cancer [93]. This method uses a domain adaptation model to align data distributions from different modalities and applies a cross-modal knowledge distillation model to predict heterogeneity across diverse patients while identifying key functional pathways and genes [93].
The integration of bulk and single-cell RNA sequencing data is particularly powerful, as it combines the statistical power and cost-effectiveness of bulk sequencing with the resolution of single-cell approaches [93]. This cross-modal integration helps researchers understand how cell-level processes contribute to population-level observations, bridging critical gaps in our understanding of complex biological systems.
Network Analysis and Pathway Integration
Cytoscape provides a robust framework for network analysis of differentially expressed genes from RNA-seq experiments [94]. The workflow involves identifying differentially expressed genes, retrieving relevant networks from public databases like STRING, integrating and visualizing experimental data, performing network functional enrichment analysis, and exporting network visualizations [94]. The STRING app within Cytoscape enables the construction of protein-protein interaction networks from gene lists and performs functional enrichment analysis including Gene Ontology, KEGG Pathways, and other biological databases [94].
This network-based approach allows researchers to move beyond simple gene lists to understand how differentially expressed genes interact within broader biological systems, identifying key regulatory nodes and functional modules that might be targeted for therapeutic intervention.
Table 2: Multi-Omics Integration Tools and Their Applications
| Tool/Platform | Primary Function | Data Types Integrated | Key Features |
|---|---|---|---|
| DeepTEX | Domain adaptation and knowledge distillation | Bulk RNA-seq, scRNA-seq | Identifies heterogeneity across patients; Predicts key functional pathways and genes |
| Cytoscape/STRING | Network analysis and visualization | RNA-seq data, protein-protein interactions | Functional enrichment analysis; Interaction network mapping; Customizable visualizations |
| Searchlight | Automated exploration and interpretation | Multiple differential comparisons | Pathway analysis; Signature analysis; Automated reporting |
Experimental Protocols and Methodologies
Standardized Sample Processing
GeneLab provides publicly available Standard Operating Procedures (SOPs) for sample extraction, library preparation, and sequencing steps using various approaches and kits [7]. These standardized protocols ensure reproducibility and comparability across studies, which is especially important in multi-omics research where data quality directly impacts integration success. For mRNA-focused libraries, the recommended sequencing depth is between 10-20 million paired-end reads when RNA quality is high (RIN > 8) [67]. For total RNA methods that capture long noncoding RNAs as well, sequencing depth of 25-60 million paired-end reads is recommended, which is also suitable for degraded RNA samples [67].
Quality Control and Normalization
Quality control begins with FastQC for assessing read quality, followed by adapter trimming using Trimmomatic [19]. Alignment to reference genomes is performed with STAR, which efficiently handles splice junctions, while HTSeq-count generates the count matrices for downstream analysis [19]. For differential expression, DESeq2 performs internal normalization using size factors that account for differences in sequencing depth between samples [19]. The method applies stringent filtering to low-count genes prior to differential expression analysis to increase statistical power [19].
For visualization purposes, particularly in Principal Component Analysis (PCA), a variance stabilizing transformation is applied to the count data before dimensionality reduction [19]. This transformation stabilizes variance across the dynamic range of expression values, preventing highly expressed genes from dominating the variance structure [19]. PCA plots typically include the 1,000 genes with the highest variance, as these contribute most to differences between samples and groups [19].
Visualization Approaches for Multi-Omics Data
Effective visualization is critical for interpreting complex multi-omics datasets. The following diagrams illustrate key workflows and relationships in bulk RNA-seq and multi-omics integration.
Bulk RNA-Seq Experimental Workflow
Multi-Omics Data Integration Framework
Differential Expression Analysis Pipeline
Table 3: Essential Research Reagents and Computational Resources for Bulk RNA-Seq and Multi-Omics Integration
| Category | Item/Resource | Function/Application |
|---|---|---|
| Wet-Lab Reagents | Poly-A Selection Kits | Enrichment of messenger RNA from total RNA samples |
| Ribo-depletion Kits | Removal of ribosomal RNA for total RNA sequencing | |
| RNA Extraction Reagents | High-quality RNA isolation maintaining integrity | |
| Library Preparation Kits | Construction of sequencing-ready libraries | |
| Computational Tools | DESeq2 | Differential gene expression analysis |
| STAR Aligner | Spliced alignment of RNA-seq reads | |
| HTSeq-count | Gene-level quantification of aligned reads | |
| Cytoscape & STRING App | Network analysis and visualization | |
| Searchlight | Automated exploration and interpretation | |
| Reference Databases | GENCODE Annotations | Comprehensive gene annotation for alignment |
| STRING Database | Protein-protein interaction networks | |
| GO, KEGG, Reactome | Pathway and functional enrichment analysis |
The integration of bulk RNA-seq with other omics data represents a powerful paradigm for systems biology, enabling researchers to construct comprehensive models of biological systems. As computational methods continue to advance, particularly in domain adaptation and knowledge distillation frameworks like DeepTEX [93], the potential for uncovering novel biological insights through multi-omics integration expands significantly. The development of automated analysis pipelines such as Searchlight [92] further accelerates this process by reducing the time and expertise required to progress from raw data to biological interpretation. As these technologies mature, standardized experimental designs [27] [67] and processing workflows [19] [7] will ensure the generation of high-quality, reproducible data that forms the foundation for robust multi-omics integration. This integrated approach promises to advance our understanding of complex biological systems and accelerate the development of novel therapeutic strategies.
The translation of innovative research assays into clinically validated diagnostic tools represents a critical pathway in modern precision medicine. Bulk RNA sequencing (RNA-seq) has emerged as a powerful research technology for comprehensive transcriptomic profiling, yet its adoption in clinical diagnostics has proceeded more cautiously than DNA-based sequencing. Clinical validation provides the essential bridge between research findings and clinically actionable diagnostics, establishing rigorous evidence of an assay's reliability, accuracy, and clinical utility for informed patient management decisions. This process demands meticulous attention to analytical performance, reproducibility, and clinical correlation across diverse patient populations.
The fundamental challenge in clinical validation of bulk RNA-seq lies in transitioning from a research-grade tool capable of discovering biologically interesting patterns to a clinically reliable test that consistently informs medical decisions. While research applications prioritize discovery, clinical diagnostics demand standardization, reproducibility, and definitive interpretation guidelines. This whitepaper examines the frameworks, methodologies, and considerations essential for translating bulk RNA sequencing from a research technology into clinically validated diagnostic applications, with particular emphasis on validation frameworks, quality metrics, and clinical implementation pathways.
Fundamental Principles and Workflow
Bulk RNA sequencing is a powerful method for transcriptomic analysis of pooled cell populations, tissue sections, or biopsies that measures the average expression level of individual genes across hundreds to millions of input cells [24]. This technology provides a global perspective on gene expression differences between sample groups, making it particularly valuable for identifying consistent transcriptional patterns associated with disease states, treatment responses, or other biological conditions. The technology's strength lies in its comprehensive capture of transcriptomic information from a population of cells, offering a composite profile that reflects the predominant biological signals within a sample.
The core workflow begins with sample preparation, where RNA is extracted from the biological specimen and converted into a sequencing-ready library. Critical pre-analytical steps include RNA quality assessment, with metrics like RNA Integrity Number (RIN) serving as crucial quality indicators [86] [10]. Library preparation involves converting RNA to complementary DNA (cDNA), fragmenting the molecules, attaching platform-specific adapters, and amplifying the library to generate sufficient material for sequencing [18]. The prepared libraries are then sequenced using next-generation sequencing (NGS) platforms, most commonly Illumina systems, which generate millions of short reads that represent fragments of the original transcript pool [18].
Analytical Outputs and Applications
Bulk RNA-seq data analysis generates multiple dimensions of transcriptomic information beyond simple gene expression quantification. The primary analytical outputs include: (1) gene expression quantification through read counts aligned to genomic features; (2) identification of differentially expressed genes between experimental conditions; (3) detection of alternative splicing events and isoform usage; (4) discovery of gene fusions resulting from chromosomal rearrangements; and (5) variant calling including single nucleotide variants and small insertions/deletions [95]. This multi-faceted data output makes bulk RNA-seq uniquely positioned to address diverse biological questions from a single assay.
In clinical research contexts, bulk RNA-seq applications span multiple domains including disease classification, biomarker discovery, therapeutic target identification, and treatment response prediction [95]. In oncology, for example, RNA-seq can reveal clinically relevant alterations such as gene fusions that may be missed by DNA-only testing [86]. In Mendelian disorders, RNA sequencing provides functional data that helps interpret variants of uncertain significance identified through DNA sequencing [96]. The technology's ability to simultaneously capture multiple types of transcriptomic alterations from a single test makes it particularly efficient for comprehensive molecular profiling.
Clinical Validation Frameworks and Standards
Comprehensive Validation Approaches
Clinical validation of bulk RNA-seq requires a multi-tiered approach that establishes analytical accuracy, clinical reproducibility, and real-world utility. A robust framework encompasses three critical phases: (1) analytical validation using reference standards and cell lines; (2) orthogonal verification using patient samples and established methods; and (3) clinical utility assessment through real-world application [86]. This comprehensive approach ensures that the test not only performs reliably under controlled conditions but also provides clinically actionable information that improves patient management.
For bulk RNA-seq specifically, validation must address the unique challenges of transcriptomic analysis, including RNA stability, sample quality variability, and the dynamic nature of gene expression. A successfully implemented framework was demonstrated in a recent study that validated a combined RNA and DNA exome assay across 2,230 clinical tumor samples [86]. The validation process utilized exome-wide somatic reference standards containing 3,042 single nucleotide variants and 47,466 copy number variations, establishing rigorous performance benchmarks across multiple sequencing runs and varying tumor purities [86]. This large-scale approach provides a template for comprehensive RNA-seq assay validation.
Regulatory and Quality Considerations
The transition of RNA-seq from research to clinical applications necessitates adherence to established regulatory frameworks and quality standards. Unlike DNA sequencing, which has benefited from standardized development and validation guidelines established through collaborative efforts among manufacturers, clinical providers, and regulatory agencies, RNA-seq lacks similar comprehensive oversight [83]. This regulatory gap presents challenges for clinical implementation, necessitating that developers of RNA-seq diagnostics establish rigorous internal standards and validation protocols.
Key considerations for clinical RNA-seq validation include establishing specimen stability metrics, determining optimal sample handling procedures, implementing batch effect controls, and defining minimum RNA quality thresholds [83] [10]. For example, preanalytical metrics including specimen collection, RNA integrity, and genomic DNA contamination have been identified as having the highest failure rates in RNA-seq workflows, necessitating additional quality controls such as secondary DNase treatment to reduce genomic DNA levels [83]. These quality measures directly impact downstream analytical performance and must be rigorously validated during test development.
Table 1: Key Components of Clinical Validation Frameworks for Bulk RNA-Seq
| Validation Phase | Key Components | Performance Metrics |
|---|---|---|
| Analytical Validation | Reference materials, cell lines, accuracy studies | Sensitivity, specificity, precision, accuracy |
| Orthogonal Verification | Patient samples, method comparison, reproducibility | Concordance rates, reproducibility, precision |
| Clinical Utility Assessment | Real-world clinical samples, outcome correlation | Clinical sensitivity/specificity, positive/negative predictive values |
Bulk RNA Sequencing in Diagnostic Applications
Oncology Diagnostics
Bulk RNA sequencing has demonstrated significant utility in oncology diagnostics, where it complements DNA-based approaches by capturing the functional transcriptomic consequences of genomic alterations. In clinical oncology, combined RNA and DNA sequencing approaches have been shown to improve the detection of clinically actionable alterations, with one large-scale study reporting the identification of such alterations in 98% of cases [86]. The integration of RNA-seq data enables direct correlation of somatic alterations with gene expression patterns, recovery of variants missed by DNA-only testing, and improved detection of gene fusions and complex genomic rearrangements [86].
Specific applications in oncology include cancer classification, biomarker discovery, and therapy selection. For example, gene fusions are well-documented as major cancer drivers, with some recurrent fusions serving as diagnostic tools, such as the RUNX1–RUNX1T1 fusion for diagnosis of acute myeloid leukemia [95]. Bulk RNA-seq enables discovery of novel gene fusions that may offer therapeutic opportunities, though challenges remain in minimizing false positives and improving detection sensitivity for clinical implementation [95]. Advanced computational approaches like the Data-Enriched Efficient PrEcise STatistical fusion detection (DEEPEST) algorithm have been developed to address these limitations, effectively minimizing false positives while improving detection sensitivity [95].
Mendelian and Rare Disorders
In Mendelian disorder diagnostics, RNA sequencing has emerged as a powerful tool for resolving ambiguous cases that remain unexplained after DNA sequencing. Despite rapid advancements in clinical sequencing, over half of diagnostic evaluations still lack definitive results, creating a significant diagnostic gap that RNA-seq can help address [96]. Clinical validation studies have demonstrated approaches where RNA sequencing tests process samples from fibroblasts or blood and derive clinical interpretations based on analytical detection of outliers in gene expression and splicing patterns [96].
The validation paradigm for rare disorder diagnostics involves establishing reference ranges for each gene and junction based on expression distributions from control data, then evaluating clinical performance using positive samples with previously identified diagnostic findings [96]. This approach was implemented in a study that developed a clinical diagnostic RNA-seq test for individuals with suspected genetic disorders who had existing or concurrent comprehensive DNA diagnostic testing [96]. The validation cohort included 130 samples (90 negative and 40 positive samples), providing statistical robustness for clinical implementation [96].
Infectious Disease and Complex Traits
Bulk RNA-seq applications extend to infectious diseases and complex traits, where transcriptomic profiling can identify host response patterns associated with disease states or outcomes. In post-COVID-19 condition (PCC), for example, bulk RNA sequencing of whole blood has identified differentially expressed genes associated with persistent symptoms, with enriched pathways related to interferon signaling and anti-viral immune processes [97]. These findings point to subtle ongoing inflammatory responses characterizing the PCC transcriptome, providing insights into potential mechanisms and therapeutic targets [97].
The experimental design for such studies typically involves careful participant phenotyping and appropriate control groups to distinguish disease-specific signals from general inflammatory patterns. In the PCC study, researchers included four groups: SARS-CoV-2 positive cases with fatigue, SARS-CoV-2 positive cases without fatigue, and SARS-CoV-2 negative cases with and without fatigue [97]. This sophisticated design enabled identification of transcriptomic features specifically associated with post-COVID condition beyond non-specific fatigue symptoms.
Experimental Protocols and Methodologies
Sample Preparation and Quality Control
Robust sample preparation and rigorous quality control form the foundation of clinically valid RNA-seq data. The preanalytical phase begins with nucleic acid isolation, with specific protocols varying by sample type. For fresh frozen solid tumors, the AllPrep DNA/RNA Mini Kit is commonly used, while for formalin-fixed paraffin-embedded (FFPE) tissues, the AllPrep DNA/RNA FFPE Kit is more appropriate [86]. For blood-based RNA sequencing, collection in specialized systems like PAXgene Blood RNA tubes followed by purification using systems like QIAsymphony PAXgene Blood RNA kit provides standardized starting material [97].
Quality assessment of extracted RNA represents a critical checkpoint before proceeding to library preparation. Essential quality metrics include RNA concentration, purity (assessed by A260/A280 and A260/A230 ratios), and integrity using measures such as RNA Integrity Number (RIN) determined by platforms like Agilent TapeStation or Bioanalyzer [86] [10]. The stringent application of RNA quality thresholds is essential, as RNA degradation significantly impacts sequencing results and can compromise clinical interpretation. Studies have demonstrated that preanalytical metrics including specimen collection, RNA integrity, and genomic DNA contamination exhibit the highest failure rates in RNA-seq workflows, necessitating careful quality monitoring at these stages [83].
Library Preparation and Sequencing
Library preparation protocols for bulk RNA-seq vary based on the specific application and sample type. For fresh frozen tissue RNA, library construction is typically performed with the TruSeq stranded mRNA kit, while for FFPE tissues, exome capture kits like SureSelect XTHS2 are often employed [86]. The selection between poly-A enrichment and rRNA depletion approaches depends on the research question and desired transcriptome coverage. Poly-A selection enriches for protein-coding mRNAs, while rRNA depletion provides broader coverage including non-coding RNAs.
The standard workflow involves several key steps: (1) reverse transcription to create cDNA from RNA; (2) fragmentation to generate appropriately sized fragments; (3) adapter ligation to add platform-specific sequences; and (4) amplification to generate sufficient material for sequencing [18]. For clinical applications, consistency in these steps is critical, as small variations can introduce biases that affect downstream results. After preparation, libraries are quantified and quality-checked before sequencing, typically on Illumina platforms like NovaSeq 6000, with target sequencing depths varying by application but generally ranging from 20-100 million reads per sample for robust transcriptional profiling [86] [97].
Bioinformatics Analysis Pipeline
The bioinformatics analysis of bulk RNA-seq data involves multiple processing steps that transform raw sequencing data into biologically interpretable results. The standard workflow begins with quality control of raw sequencing files using tools like FastQC to assess read quality, adapter contamination, and other potential issues [19] [18]. Following quality assessment, adapter trimming and quality filtering are performed using tools like Trimmomatic or Cutadapt to remove low-quality bases and adapter sequences [18].
Processed reads are then aligned to a reference genome using splice-aware aligners such as STAR, which efficiently handles reads spanning exon-exon junctions [86] [21]. Following alignment, gene-level quantification assigns reads to genomic features using tools like featureCounts or HTSeq-count, generating a count matrix that represents the expression level of each gene in each sample [19] [18]. For clinical applications, alignment is typically performed against standard references like GRCh38 for human samples, with careful attention to potential confounding factors such as genomic DNA contamination [21].
Downstream analysis includes normalization to account for technical variations in library size and composition, followed by differential expression analysis using statistical methods implemented in tools like DESeq2 or edgeR [19] [97]. These tools apply appropriate statistical models to identify genes showing significant expression differences between experimental conditions, while controlling for multiple testing using methods like Benjamini-Hochberg false discovery rate correction [19]. Additional analyses may include pathway enrichment, immune cell deconvolution, and visualization through PCA plots, heatmaps, and volcano plots [19] [97].
Quality Control and Validation Metrics
Multi-Layered QC Framework
Implementing a comprehensive quality control framework throughout the RNA-seq workflow is essential for generating clinically reliable data. An effective approach employs multilayered quality metrics across preanalytical, analytical, and postanalytical processes [83]. This framework integrates established internal practices with validated best practices to ensure accurate results and reliable interpretation, particularly for large RNA-seq datasets where batch effects and technical variability can obscure biological signals.
The preanalytical phase requires special attention, as variables at this stage significantly impact downstream results. Key preanalytical considerations include specimen collection methods, RNA stabilization, storage conditions, and extraction consistency [83] [10]. For blood-based RNA sequencing, for example, collection in specialized systems like PAXgene Blood RNA tubes followed by strict adherence to processing protocols helps maintain RNA integrity and minimize artifactual changes in gene expression [97]. Additional preanalytical steps such as secondary DNase treatment have been shown to significantly reduce genomic DNA contamination, lowering intergenic read alignment and improving data quality [83].
Analytical Performance Metrics
Clinical validation of bulk RNA-seq requires establishing and monitoring specific analytical performance metrics that demonstrate assay reliability. Key metrics include sensitivity (the ability to detect true positives), specificity (the ability to avoid false positives), precision (reproducibility across replicates and runs), and accuracy (agreement with reference methods) [86]. These metrics should be established for each application, whether detecting differentially expressed genes, identifying splice variants, or discovering gene fusions.
For gene expression quantification, validation should establish dynamic range (the span of expression levels accurately quantified), limit of detection (the lowest expression level reliably distinguished from background), and precision (reproducibility across technical replicates) [86] [83]. In one large-scale validation study, researchers utilized custom reference samples containing 3,042 single nucleotide variants and 47,466 copy number variations to establish analytical performance across multiple sequencing runs and varying tumor purities [86]. This approach provides a template for comprehensive analytical validation of RNA-seq assays.
Table 2: Essential Quality Control Metrics for Clinical RNA-Seq
| Process Stage | QC Metric | Target Threshold | Clinical Significance |
|---|---|---|---|
| Sample QC | RNA Integrity Number (RIN) | >7.0 [10] | Preserved transcriptome representation |
| DNA Contamination | Absence of gDNA bands | Avoids spurious reads and quantification errors | |
| Sequencing QC | Q30 Score | >90% [86] | High base calling accuracy |
| Mapping Rate | >70% | Efficient read utilization | |
| Analysis QC | Gene Detection | Expected range for tissue | Sufficient transcriptome coverage |
| Housekeeping Genes | Stable expression | Sample quality verification |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Clinical RNA-Seq Applications
| Reagent/Category | Specific Examples | Function in Workflow |
|---|---|---|
| RNA Isolation Kits | AllPrep DNA/RNA Mini Kit (Qiagen) [86] | Simultaneous DNA/RNA extraction from fresh frozen tissues |
| AllPrep DNA/RNA FFPE Kit (Qiagen) [86] | Nucleic acid extraction from formalin-fixed tissues | |
| PAXgene Blood RNA Kit (PreAnalytiX) [97] | Stabilization and extraction from whole blood | |
| Library Preparation | TruSeq stranded mRNA kit (Illumina) [86] | mRNA library construction from high-quality RNA |
| SureSelect XTHS2 (Agilent) [86] | Target enrichment for degraded samples (FFPE) | |
| NEBNext Poly(A) mRNA Isolation [10] | mRNA enrichment for directional libraries | |
| Quality Assessment | Qubit RNA HS Assay (Thermo Fisher) [86] | Accurate RNA quantification |
| Agilent TapeStation/Bioanalyzer [86] [10] | RNA integrity assessment (RIN) | |
| Enzymes & Amplification | SuperScript Reverse Transcriptase [21] | cDNA synthesis from RNA templates |
| KAPA HiFi HotStart ReadyMix [21] | High-fidelity library amplification |
Data Analysis and Interpretation Framework
Statistical Considerations for Clinical Applications
Robust statistical analysis forms the cornerstone of clinically valid RNA-seq data interpretation. For differential expression analysis, methods like those implemented in DESeq2 are widely used, employing a negative binomial distribution to model count data and the Wald test to assess statistical significance [19]. These approaches account for the inherent variability in RNA-seq data while controlling for multiple testing through false discovery rate (FDR) corrections [19] [97]. The threshold for statistical significance must be established based on the clinical context, with more stringent thresholds often required for diagnostic applications compared to exploratory research.
Beyond identifying differentially expressed genes, effect size estimation using empirical Bayes shrinkage methods provides more reliable fold-change estimates, particularly for low-count genes [19]. These methods, implemented in packages like apeglm, help prevent technical artifacts from inflating fold-change estimates and provide additional confidence metrics such as s-values that indicate confidence in the direction of expression changes [19]. For clinical applications, both statistical significance and effect size should be considered when interpreting the biological and clinical relevance of findings.
Clinical Interpretation and Reporting
Translating RNA-seq results into clinically actionable information requires careful interpretation within the relevant biological and clinical context. Interpretation frameworks should integrate multiple lines of evidence, including the magnitude of expression changes, known biological pathways, and prior clinical knowledge about specific genes or signatures [86] [96]. For example, in Mendelian disorder diagnostics, interpretation is based on detecting outliers in gene expression and splicing patterns compared to established reference ranges [96].
Effective clinical reporting should clearly distinguish between well-established findings and those with emerging evidence, providing clinicians with appropriate context for decision-making. Reports should include information about assay limitations, sample quality, and the confidence of specific findings [86] [83]. For complex results such as gene expression signatures, visualization techniques like heatmaps and principal component analysis plots can help communicate patterns effectively to clinical stakeholders [19] [10].
The clinical validation of bulk RNA sequencing represents a methodical process that transforms a powerful research tool into a reliable clinical diagnostic. This transition requires rigorous analytical validation, orthogonal verification, and demonstrated clinical utility across appropriately sized patient cohorts. As validation frameworks mature and standards emerge, bulk RNA-seq is poised to expand its role in clinical diagnostics, particularly in applications that benefit from its comprehensive capture of transcriptomic information. The continued refinement of analytical methods, quality control processes, and interpretation guidelines will further strengthen the clinical implementation of this versatile technology, ultimately enhancing patient care through more precise molecular diagnostics.
Bulk RNA sequencing (Bulk RNA-Seq) stands as a foundational methodology in modern molecular biology, providing critical insights into gene expression patterns across diverse biological systems. This technique measures the average expression level of individual genes from samples consisting of pooled cell populations, tissue sections, or biopsies, delivering a global perspective on transcriptional activity [24]. Unlike emerging single-cell approaches, bulk RNA-seq generates consolidated expression profiles representing hundreds to millions of input cells, making it indispensable for capturing overall transcriptional differences between experimental conditions, disease states, or treatment responses [10] [98].
The fundamental value of bulk RNA-seq lies in its powerful capacity for large-scale gene expression profiling, enabling comparisons between different conditions (e.g., healthy vs. diseased or treated vs. untreated), discovery of novel transcripts, biomarker identification, and comprehensive pathway analyses [7]. While it provides broad, quantitative profiling capabilities, the method averages signals across many cells, which means it cannot resolve single-cell differences—a limitation addressed by complementary single-cell technologies [99]. Despite this limitation, bulk RNA-seq remains widely deployed due to its cost-effectiveness, established analytical frameworks, and proven utility across countless biomedical research contexts.
The core process involves converting RNA molecules into complementary DNA (cDNA) and sequencing them using next-generation sequencing platforms. Because ribosomal RNA (rRNA) constitutes more than 80% of total RNA and is typically not the analytical focus, it is removed during sample preparation prior to cDNA conversion, either through ribo-depletion or by selecting for messenger RNA (mRNA) using polyA-selection [7]. The resulting data empowers researchers to answer fundamental questions about transcriptional regulation in development, disease, and treatment response.
Core Technologies and Methodological Foundations
Experimental Design and Sequencing Standards
Robust experimental design forms the critical foundation for generating biologically meaningful bulk RNA-seq data. The ENCODE consortium has established comprehensive standards to ensure data quality and reproducibility. According to these guidelines, bulk RNA-seq experiments should ideally include two or more biological replicates to account for natural variation, with exemptions granted only for exceptional circumstances such as limited material availability [100]. Each replicate should target 20-30 million aligned reads, though specific applications like shRNA knockdown experiments may require only 10 million aligned reads [100].
Sequencing read length should meet a minimum of 50 base pairs, with paired-end sequencing strongly recommended over single-end layouts because paired-end reads provide more robust expression estimates at effectively the same cost per base [100] [6]. Replicate concordance represents another crucial quality metric, with gene-level quantifications expected to demonstrate a Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates [100]. Strategic batch effect mitigation is essential throughout experimentation, library preparation, and sequencing runs to prevent technical artifacts from masquerading as biological findings [10].
Table 1: Key Experimental Standards for Bulk RNA-Seq
| Experimental Parameter | Standard Specification | Quality Metric |
|---|---|---|
| Biological Replicates | ≥2 replicates | Spearman correlation >0.9 (isogenic) |
| Sequencing Depth | 20-30 million aligned reads | >0.8 (anisogenic) |
| Read Length | Minimum 50 bp | Library insert size >200 bp |
| Spike-in Controls | ERCC Spike-ins | ~2% of final mapped reads |
| Library Type | Paired-end recommended | Strand-specific or non-specific |
Library Preparation and Quality Control
Library preparation transforms RNA samples into sequenceable libraries through a multi-step process. Beginning with RNA extraction, samples undergo pre-experimental quality controls including concentration determination with Qubit and normalization to minimize read variability during sequencing [24]. For mRNA sequencing, poly(A)+ RNA selection is performed using oligo(dT) primers or rRNA depletion to remove abundant ribosomal RNA [10]. Reverse transcription follows, converting RNA to complementary DNA (cDNA), with unique barcodes (indexes) added to each sample to enable multiplexing—pooling multiple samples for simultaneous sequencing [24] [101].
The final library preparation step generates a sequenceable cDNA library, with quality and concentration determined using systems like the Agilent TapeStation [24]. Throughout this process, careful attention to potential batch effects is crucial, including minimizing users, processing controls and experimental conditions simultaneously, and maintaining consistent RNA isolation procedures [10]. For samples with limited starting material, it may not be possible to perform all quality controls due to low RNA yield, requiring adjustments to standard protocols [24].
Computational Processing and Analysis Pipeline
Computational processing of bulk RNA-seq data involves multiple sophisticated steps to transform raw sequencing reads into interpretable gene expression data. The ENCODE4 bulk RNA-seq pipeline represents a standardized approach that accepts FASTQ files as input and performs alignment, generates signal tracks, and quantifies genes and isoforms [100]. This pipeline accommodates both replicated and unreplicated, paired-end or single-end, and strand-specific or non-strand specific RNA-seq libraries, providing comprehensive analytical capabilities.
A prominent alternative is the nf-core RNA-seq workflow, which automates the entire processing pipeline from raw reads to count matrices [6]. This workflow utilizes the "STAR-salmon" option, performing spliced alignment to the genome with STAR, projecting those alignments onto the transcriptome, and conducting alignment-based quantification with Salmon. This integrated approach provides both comprehensive quality control metrics and statistically robust expression estimates.
Table 2: Core Computational Tools for Bulk RNA-Seq Analysis
| Analytical Step | Recommended Tools | Primary Function |
|---|---|---|
| Read Alignment | STAR [60] [100] [6] | Splice-aware alignment to genome |
| Gene Quantification | featureCounts [60], RSEM [100] | Read counting for genes |
| Transcript Quantification | Salmon [100] [6], kallisto [100] | Pseudoalignment for isoform-level counts |
| Differential Expression | DESeq2 [60], limma [6], edgeR [10] | Statistical testing for expression changes |
| Quality Control | FastQC, MultiQC | Sequence quality assessment |
The following workflow diagram illustrates the integrated bulk RNA-seq analysis process, combining experimental and computational components:
Established Analytical Frameworks
Read Alignment and Expression Quantification
The initial computational phase involves determining the genomic origins of sequencing reads, addressing two levels of uncertainty: identifying the most likely transcript of origin for each read, and converting read assignments to counts while modeling inherent assignment uncertainty [6]. Two predominant approaches have emerged for this task, each with distinct advantages.
Alignment-based quantification utilizes splice-aware aligners like STAR to map reads directly to a reference genome, accommodating alignment gaps due to introns through specialized algorithms [60] [6]. The resulting BAM files containing alignment coordinates then undergo quantification using tools like featureCounts to generate gene-level counts, or RSEM to estimate expression at both gene and isoform levels [60] [100]. This approach preserves detailed alignment information valuable for quality control and alternative splicing analyses.
Pseudoalignment-based quantification, implemented in tools like Salmon and kallisto, offers a computationally efficient alternative by using substring matching to probabilistically determine transcript origins without base-level alignment [6]. This approach simultaneously addresses both levels of quantification uncertainty and produces sample-level counts that can be aggregated into count matrices. Due to its speed and statistical robustness, pseudoalignment has gained popularity, particularly for large-scale studies.
For comprehensive analysis, a hybrid approach is often recommended: using STAR for initial alignment to enable quality control metrics generation, followed by Salmon in alignment-based mode to leverage its sophisticated statistical models for count estimation [6]. This strategy combines the QC benefits of alignment with the quantification advantages of pseudoalignment.
Differential Expression Analysis
Differential expression analysis represents a cornerstone of bulk RNA-seq investigations, identifying genes with statistically significant expression changes between experimental conditions. The tool landscape includes well-established packages like DESeq2, limma, and edgeR, each employing distinct statistical frameworks for this purpose [60] [6] [10].
DESeq2 utilizes a negative binomial generalized linear model to test for differential expression, incorporating data-driven prior distributions for dispersion estimation and fold change shrinkage to improve stability and interpretability of results [60]. The limma package employs a linear modeling framework combined with empirical Bayes moderation of standard errors, providing robust performance across diverse experimental designs [6]. edgeR similarly uses a negative binomial model but with different estimation approaches for dispersion and statistical testing [10].
The analytical process typically begins with quality assessment using principal component analysis (PCA) to visualize sample-to-sample distances and identify potential outliers or batch effects [10]. Following model fitting and statistical testing, results undergo multiple testing correction (e.g., Benjamini-Hochberg procedure) to control false discovery rates. Visualization through volcano plots, MA plots, and heatmaps facilitates interpretation of the results, highlighting both individual gene changes and global expression patterns.
Advanced Applications and Specialized Analyses
Beyond standard gene expression profiling, bulk RNA-seq supports specialized applications that expand its utility in biomedical research. Transposable element analysis can be performed using tools like TEcount from the TEToolkit, enabling quantification of TE subfamily expression by allowing multi-mapping reads across repetitive genomic regions [60]. This approach has revealed important roles for evolutionary young L1 elements in various biological processes.
Strand-specific analysis enables differentiation between sense and antisense transcription, providing insights into regulatory mechanisms involving antisense RNAs. This requires specialized processing during alignment and quantification, preserving strand information through parameters like -s 2 in featureCounts [60]. The resulting strand-specific signals can be visualized over genomic features like transposable elements to elucidate their transcriptional regulation.
Pathway and enrichment analyses move beyond individual genes to identify coordinated biological processes, using methods like over-representation analysis (ORA) with Gene Ontology (GO) or KEGG databases, and upstream regulator analysis (URA) to infer transcription factor activities [92]. These approaches help place differential expression results in broader biological context, revealing underlying mechanisms and functional consequences.
Emerging Applications in Biomedicine
Biomarker Discovery and Clinical Translation
Bulk RNA-seq has emerged as a powerful platform for biomarker discovery, generating molecular signatures with diagnostic, prognostic, and predictive potential across diverse disease areas. In cancer research, transcriptomic profiling has identified expression signatures that stratify patients into molecular subtypes with distinct clinical outcomes and treatment responses, enabling more personalized therapeutic approaches. The technology's comprehensive nature allows for unbiased discovery of novel biomarkers beyond candidate genes, capturing pathway activities and biological processes relevant to disease mechanisms.
The implementation of standardized processing pipelines, such as the GeneLab consensus pipeline for identifying spaceflight-induced differentially expressed genes, demonstrates how systematic analytical approaches enhance reproducibility and reliability of biomarker signatures [7]. As the field advances, integration of bulk RNA-seq data with other molecular profiling data (genomic, proteomic) and clinical information will further strengthen biomarker development, creating multidimensional models of disease states and treatment responses.
Drug Development Applications
In pharmaceutical research and development, bulk RNA-seq provides critical insights throughout the drug discovery pipeline. During target identification and validation, transcriptomic profiling can reveal disease-associated genes and pathways, prioritizing targets with strong biological rationale. In preclinical development, RNA-seq analyses of model systems treated with candidate compounds help elucidate mechanisms of action, identify potential toxicity concerns, and discover pharmacodynamic biomarkers for use in early clinical trials.
Specialized applications include shRNA and CRISPR screening followed by RNA-seq, which have specific standards including 10 million aligned reads per replicate and verification of knockdown efficiency relative to controls [100]. These functional genomics approaches enable systematic identification of genes that modulate disease-relevant phenotypes or drug responses. As the technology continues to evolve, its integration with high-throughput screening platforms and sophisticated computational models promises to accelerate therapeutic development.
Integration with Multi-Omic Approaches
The evolving landscape of biomedical research increasingly emphasizes multi-omic integration, combining transcriptomic data with other molecular measurements to build comprehensive models of biological systems. Bulk RNA-seq serves as a fundamental component in these integrated approaches, contributing crucial information about the functional genomic state that links genetic variation, epigenetic regulation, and phenotypic outcomes.
Advanced analytical frameworks now enable combined analysis of bulk RNA-seq data with genomic, epigenomic, and proteomic datasets, revealing coordinated molecular changes across regulatory layers. While single-cell multi-omic technologies are rapidly advancing, bulk approaches remain valuable for profiling large sample cohorts with deep sequencing coverage, particularly when combined with deconvolution methods that estimate cellular composition from transcriptomic data. This integration across platforms and molecular layers represents a powerful strategy for unraveling complex biological systems and disease processes.
Successful bulk RNA-seq experiments rely on carefully selected reagents and reference materials that ensure experimental quality and reproducibility. The following table catalogizes key solutions utilized throughout standard workflows:
Table 3: Essential Research Reagent Solutions for Bulk RNA-Seq
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| RNA Extraction Kits | PicoPure RNA Isolation Kit [10] | RNA purification from cells/tissues |
| mRNA Enrichment | NEBNext Poly(A) mRNA magnetic isolation kits [10] | Selection of polyadenylated transcripts |
| Library Preparation | NEBNext Ultra DNA Library Prep Kit [10] | cDNA library construction for sequencing |
| Spike-in Controls | ERCC Spike-in Mix (Ambion) [100] | Technical normalization control |
| Unique Dual Indexes | CEL-seq2-type barcodes [24] | Sample multiplexing and demultiplexing |
| Reference Genomes | GRCh38, mm10 with GENCODE annotations [60] [100] | Read alignment and quantification baseline |
| Quality Control | Agilent TapeStation [24], RNA Integrity Number (RIN) [10] | Assessment of RNA and library quality |
The strategic implementation of these reagent systems ensures robust technical performance throughout the bulk RNA-seq workflow. Spike-in controls, particularly the ERCC RNA Spike-In Mix, are essential for distinguishing technical variability from biological differences by adding known concentrations of exogenous RNA transcripts to each sample prior to library preparation [100]. Quality control metrics like RNA Integrity Number (RIN) values greater than 7.0 provide critical assessment of sample quality, with degradation potentially compromising downstream results [10].
Reference materials continue to evolve in sophistication, with current standards utilizing GRCh38 (human) or mm10 (mouse) genome assemblies with GENCODE annotations (V29 for human, M21 for mouse) to ensure accurate alignment and quantification [100]. The availability of comprehensive, well-annotated reference datasets enables more precise transcript quantification and interpretation, forming the foundation for biologically meaningful conclusions.
Visualization and Interpretation Frameworks
Automated Analysis and Visualization Platforms
The complexity of bulk RNA-seq data interpretation has motivated development of automated analysis platforms that streamline exploration and visualization. Searchlight represents one such tool that automates the essential process where biology is explored, visualized and interpreted (EVI) following data processing [92]. This freely available pipeline provides comprehensive statistical and visual analysis at global, pathway, and single-gene levels through three complementary workflows: Normalized Expression (NE), Differential Expression (DE), and Multiple Differential Expression (MDE).
The Normalized Expression workflow focuses on quality control and experimental overview, including expression distribution analysis, principal component analysis (PCA), distance analysis, and highly expressed gene analysis [92]. The Differential Expression workflow explores single comparisons between two conditions through MA plots, volcano plots, significant gene heatmaps, and pathway analyses including over-representation analysis and upstream regulator analysis. The Multiple Differential Expression workflow examines relationships between multiple differential comparisons through signature analyses, overlap statistics, and fold-change comparisons.
Commercial tools such as Ingenuity Pathway Analysis (IPA) and Partek Flow offer alternative platforms with comprehensive visualization capabilities, though Searchlight provides a freely available alternative that generates R scripts for each plot, enabling custom modification and ensuring compatibility with bioinformaticians' standard working practices [92]. These automated platforms significantly reduce the time and effort required to progress from processed data to manuscript-quality figures, accelerating the research cycle.
Advanced Visualization Strategies
Effective visualization is critical for interpreting complex bulk RNA-seq datasets and communicating biological insights. Beyond standard representations like PCA plots and heatmaps, advanced strategies enable deeper exploration of transcriptional patterns. Volcano plots simultaneously display statistical significance (-log10(p-value)) versus magnitude of change (log2(fold-change)), allowing rapid identification of the most biologically meaningful differential expressions [92]. MA plots (log-ratio versus mean average) visualize differences relative to average expression levels, highlighting potential dependence of variance on expression magnitude.
For time-series or multi-condition experiments, clustered heatmaps with dendrograms reveal coherent expression patterns across sample groups, while violin plots provide detailed representation of expression distribution for individual genes across conditions [92]. Pathway enrichment networks visualize relationships between significantly enriched biological processes, placing results in broader functional context. Specialized visualizations like genome browser tracks enable investigation of expression patterns across genomic loci, particularly valuable for studying isoform usage, regulatory elements, and non-coding transcripts.
The following diagram illustrates the core visualization framework for differential expression analysis:
Future Directions and Concluding Perspectives
As bulk RNA-seq continues to evolve, several emerging trends promise to expand its capabilities and applications in biomedical research. Integration with single-cell approaches represents a powerful future direction, where bulk sequencing provides deep coverage for confident isoform quantification and detection of low-abundance transcripts, while single-cell methods resolve cellular heterogeneity. This complementary approach leverages the respective strengths of each technology, enabling comprehensive understanding of both population-level and cell-type-specific transcriptional regulation.
Multi-omic integration constitutes another significant frontier, with computational methods advancing to combine bulk RNA-seq data with epigenomic, proteomic, and metabolomic measurements. These integrated analyses provide more comprehensive views of biological systems, revealing how molecular regulation at one level influences other cellular components. As spatial transcriptomics technologies mature, integration with bulk RNA-seq will further enhance our ability to contextualize gene expression within tissue architecture and cellular neighborhoods.
Methodological refinements continue to improve the technology's performance and applicability. Computational deconvolution approaches are increasingly sophisticated, enabling estimation of cell-type composition and cell-type-specific expression from bulk data, thereby recovering some of the resolution traditionally associated only with single-cell methods. Advancements in long-read sequencing technologies promise to revolutionize isoform-level analysis, providing more accurate characterization of transcript diversity when combined with standard short-read bulk approaches.
In conclusion, bulk RNA-seq remains an indispensable tool in biomedical research, providing robust, cost-effective transcriptional profiling that continues to yield fundamental insights into gene regulation in health and disease. While emerging single-cell technologies capture attention for their resolution of cellular heterogeneity, bulk approaches offer complementary advantages in throughput, sensitivity, and established analytical frameworks. The future of transcriptional profiling lies not in exclusive adoption of any single approach, but in strategic integration of bulk, single-cell, and spatial methods—each contributing unique perspectives to build comprehensive understanding of biological systems. As these technologies evolve in tandem, they will collectively empower researchers to address increasingly complex biological questions and accelerate translation of genomic discoveries into clinical applications.
Conclusion
Bulk RNA sequencing remains a powerful, cost-effective tool for comprehensive transcriptome analysis, providing critical insights into gene expression patterns underlying disease mechanisms and treatment responses. Its established workflow—from careful experimental design through sophisticated bioinformatics analysis—delivers robust data for identifying differentially expressed genes and pathways. While newer single-cell technologies offer higher resolution, bulk RNA-seq continues to excel in clinical applications where population-level expression patterns are most relevant. Future advancements will likely focus on integrating bulk RNA-seq with other omics data, refining analytical pipelines for specific biological contexts, and expanding its role in personalized medicine through improved biomarker discovery and therapeutic targeting. For researchers and drug development professionals, mastering bulk RNA-seq principles and applications provides a fundamental skill set for driving innovation in biomedical research and clinical practice.
References
- 1. Sanger Sequencing vs. Next-Generation Sequencing (NGS) [https://www.genscript.com/gene-news/sanger-sequencing-vs-next-generation-sequencing.html]
- 2. Sanger Sequencing vs. Next-Generation Sequencing (NGS) [https://www.cd-genomics.com/resource-sanger-sequencing-vs-next-generation-sequencing.html]
- 3. RNA-Seq: a revolutionary tool for transcriptomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2949280/]
- 4. NGS vs Sanger Sequencing [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-sanger.html]
- 5. RNA Sequencing (RNA-Seq) Methods for NGS [https://www.thermofisher.com/us/en/home/life-science/sequencing/sequencing-learning-center/next-generation-sequencing-information/ngs-basics/rna-sequencing-methods.html]
- 6. [Tutorial] Bulk RNA-seq DE analysis [https://informatics.fas.harvard.edu/resources/tutorials/differential-expression-analysis/]
- 7. Bulk RNA Sequencing (RNA-seq) [https://science.nasa.gov/biological-physical/data/osdr/bulk-rna-sequencing-rna-seq/]
- 8. Bulk vs. single cell RNA-seq: Experimental differences and ... [https://www.10xgenomics.com/blog/bulk-vs-single-cell-RNA-seq-experimental-differences-and-advantages-of-single-cell-resolution]
- 9. A Guide to Basic RNA Sequencing Data Processing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067304/]
- 10. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 11. Bulk RNA-seq Data Standards and Processing Pipeline [https://www.encodeproject.org/data-standards/rna-seq/long-rnas/]
- 12. RNA Sequencing | RNA-Seq methods & workflows [https://www.illumina.com/techniques/sequencing/rna-sequencing.html]
- 13. RNA-Seq Experimental Design Guide for Drug Discovery [https://www.lexogen.com/blog/planning-for-success-a-strategic-design-guide-for-rna-seq-experiments-in-drug-discovery/]
- 14. Replicability of bulk RNA-Seq differential expression and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011630]
- 15. A comprehensive workflow for optimizing RNA-seq data analysis [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10414-y]
- 16. Transcriptome size matters for single-cell RNA-seq ... [https://www.nature.com/articles/s41467-025-56623-1]
- 17. A resource for whole-body gene expression map of human ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03616-4]
- 18. The Beginner's guide to bulk RNA-Seq Analysis [https://silicogene.com/blog/bulk-rna-seq-data-analysis/]
- 19. Bulk RNAseq Methodology and Analysis Guide [https://www.cores.emory.edu/eicc/_includes/documents/sections/resources/RNAseq_Methodology.html]
- 20. Introduction to Bulk RNAseq data analysis [https://bioinformatics-core-shared-training.github.io/Bulk_RNAseq_Course_Nov22/Bulk_RNAseq_Course_Base/Markdowns/07_Data_Exploration.html]
- 21. Protocol for bulk RNA sequencing of enriched human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9946790/]
- 22. Introduction to Next-Generation RNA Sequencing [https://www.lexogen.com/blog/rna-lexicon-introduction-to-next-generation-rna-sequencing/]
- 23. Prime-seq, efficient and powerful bulk RNA sequencing [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02660-8]
- 24. What is Bulk RNA sequencing (Bulk RNA-seq)? [https://www.scdiscoveries.com/support/what-is-bulk-rna-sequencing/]
- 25. What is Bulk RNA Sequencing? [https://www.cd-genomics.com/resource-bulk-rna.html]
- 26. Perspectives on Bulk-Tissue RNA Sequencing and Single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11285642/]
- 27. Experimental design considerations | Introduction to RNA-seq ... [https://hbctraining.github.io/Intro-to-rnaseq-fasrc-salmon-flipped/lessons/02_experimental_planning_considerations.html]
- 28. Sample preparation for RNA sequencing [https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/rna-extraction/rna-applications/rna-sequencing-sample-preparation.html]
- 29. Deciphering RNA-seq Library Preparation: From Principles ... [https://www.cd-genomics.com/resouce-rna-seq-library-preparation-principles-protocol.html]
- 30. RNA Library Preparation [https://www.illumina.com/techniques/sequencing/ngs-library-prep/rna.html]
- 31. NovaSeq 6000 Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html]
- 32. NovaSeq X Series specifications [https://www.illumina.com/systems/sequencing-platforms/novaseq-x-plus/specifications.html]
- 33. NovaSeq 6000 Sequencing System [https://www.illumina.com/systems/sequencing-platforms/novaseq.html]
- 34. Commentary: a review of technical considerations for planning ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12094-8]
- 35. The Complete Guide to NGS Data Types and Formats [https://ngs101.com/the-complete-guide-to-ngs-data-types-and-formats-from-raw-reads-to-analysis-ready-files/]
- 36. Evaluation of sequencing reads at scale using rdeval - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838479/]
- 37. 2025 Trends: NGS [https://www.genengnews.com/topics/genome-editing/2025-trends-ngs/]
- 38. Bulk RNA-Seq, Like a Fine Wine, Is Only Getting Better ... [https://www.viascientific.com/blogs/bulk-rna-seq-like-a-fine-wine-is-only-getting-better-with-age]
- 39. RnaXtract – a tool for extracting gene expression, variants ... [https://www.rna-seqblog.com/rnaxtract-a-tool-for-extracting-gene-expression-variants-and-cell-type-composition-from-bulk-rna-sequencing/]
- 40. Bioinformatics Workflow of RNA-Seq [https://www.cd-genomics.com/resourse-bioinformatics-workflow-of-rna-seq.html]
- 41. Differential Expression Analysis for Bulk RNA Sequencing [https://bioinformatics.ccr.cancer.gov/docs/bioinformatics-for-beginners-2025/Module2_RNA_Sequencing/Lesson14/]
- 42. Analyzing Bulk RNA-Seq Data Using CHIRP Pipeline [https://www.rna-seqblog.com/tutorial-analyzing-bulk-rna-seq-data-using-chirp-pipeline/]
- 43. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 44. Reference-based RNA-Seq data analysis [https://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/ref-based/tutorial.html]
- 45. How to Analyze RNAseq Data for Absolute Beginners Part 20 [https://ngs101.com/how-to-analyze-rnaseq-data-for-absolute-beginners-part-20-comparing-limma-deseq2-and-edger-in-differential-expression-analysis/]
- 46. Differential expression analysis with inmoose, the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183803/]
- 47. A Quick Start Guide to RNA-Seq Data Analysis [https://blog.genewiz.com/a-quick-start-guide-to-rna-seq-data-analysis]
- 48. DESeq2: An Overview of a Popular RNA-Seq Analy... [https://pluto.bio/resources/Learning%20Series/deseq2-an-overview-of-popular-rna-seq-analysis-package]
- 49. Functional Analysis for RNA-seq | Introduction to DGE [https://hbctraining.github.io/DGE_workshop_salmon/lessons/functional_analysis_2019.html]
- 50. Functional Insights Through Gene Ontology, Disease ... [https://pubmed.ncbi.nlm.nih.gov/40455152/]
- 51. 18. Gene set enrichment and pathway analysis [https://www.sc-best-practices.org/conditions/gsea_pathway.html]
- 52. GSEA [https://www.gsea-msigdb.org/]
- 53. Integrated analysis of single-cell and bulk RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10401466/]
- 54. Single‐Cell and Bulk RNA Sequencing Highlights Intra‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12379737/]
- 55. Bioinformatics perspectives on transcriptomics [https://www.rna-seqblog.com/bioinformatics-perspectives-on-transcriptomics-a-comprehensive-review-of-bulk-and-single-cell-rna-sequencing-analyses/]
- 56. Potential application of the bulk RNA sequencing in routine ... [https://pubmed.ncbi.nlm.nih.gov/40264064/?utm_source=Chrome&utm_medium=rss&utm_campaign=None&utm_content=1nASi37tjavVgwsXmk5YUf_GRBPtoGVcaaaxKnz2ejNfc5MMoY&fc=None&ff=20250423105952&v=2.18.0.post9+e462414]
- 57. Augmenting precision medicine via targeted RNA-Seq ... [https://www.nature.com/articles/s41698-025-00993-8]
- 58. Introduction to Bulk RNAseq data analysis [https://bioinformatics-core-shared-training.github.io/cruk-summer-school-2021/RNAseq/Markdowns/07_Data_Exploration.html]
- 59. Bulk RNA-sequencing pipeline and differential gene ... - Erick Lu [https://erilu.github.io/bulk-rnaseq-analysis/]
- 60. Bulk RNA sequencing analysis [https://www.protocols.io/view/bulk-rna-sequencing-analysis-cqgkvtuw.html]
- 61. Tutorials in Genomics & Bioinformatics: RNA-Seq Analysis 2025 [https://meetings.cshl.edu/courses.aspx?course=TGAC2]
- 62. Optimized murine sample sizes for RNA sequencing ... [https://www.nature.com/articles/s41467-025-65022-5]
- 63. Statistical Power Analysis for Designing Bulk, Single-Cell, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9952882/]
- 64. Public RNA-Seq and scRNA-Seq Databases [https://bigomics.ch/blog/ultimate-guide-to-public-rnaseq-and-sc-rna-seq-databases/]
- 65. Highly effective batch effect correction method for RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11718288/]
- 66. Assessing and mitigating batch effects in large-scale omics ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03401-9]
- 67. Experimental Design: Best Practices - Bioinformatics [https://bioinformatics.ccr.cancer.gov/ccbr/project-support/experimental-design-best-practices/]
- 68. Highly effective batch effect correction method for RNA-seq ... [https://www.sciencedirect.com/science/article/pii/S200103702400432X]
- 69. Stop Wasting Time: Correcting Batch Effects in Multi-Omics ... [https://pluto.bio/resources/Learning%20Series/batch-effects-multi-omics-data-analysis]
- 70. RNA integrity number [https://en.wikipedia.org/wiki/RNA_integrity_number]
- 71. What is the RNA Integrity Number (RIN)? [https://www.cd-genomics.com/longseq/resource-rna-integrity-number.html]
- 72. Essential RNA-seq Metrics: Part 1 [https://www.rna-seqblog.com/essential-rna-seq-metrics-part-1/]
- 73. An Overview On RNA Sequence Analysis [https://www.goldbio.com/blogs/articles/overview-rna-sequence-analysis?srsltid=AfmBOor_I1C6Ozd3SnVeEX2DPB8VpUqBK4Azt8jGREbNmTj2zv1Cbx00]
- 74. Commentary: a review of technical considerations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522984/]
- 75. Bulk RNA sequencing for analysis of post COVID-19 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10964710/]
- 76. a user-friendly interface for seamless dual and bulk -Seq analysis RNA [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1696823/full]
- 77. Potential application of the bulk in routine MPN... RNA sequencing [https://pubmed.ncbi.nlm.nih.gov/40264064/]
- 78. Bioinformatics Pipeline For Optimization Techniques [https://www.meegle.com/en_us/topics/bioinformatics-pipeline/bioinformatics-pipeline-for-optimization-techniques]
- 79. Quantum Algorithm Targets Metabolic Networks in Step ... [https://thequantuminsider.com/2025/11/25/quantum-algorithm-targets-metabolic-networks-in-step-toward-biological-modeling/]
- 80. Challenges and Considerations in Bulk RNA-seq [https://alitheagenomics.com/blog/challenges-and-considerations-in-bulk-rna-seq]
- 81. Integration of Bulk RNA-seq Pipeline Metrics for Assessing ... [https://pubmed.ncbi.nlm.nih.gov/40630536/]
- 82. a new approach to understanding nascent and mature mRNA [https://www.rna-seqblog.com/unraveling-rna-sequencing-challenges-a-new-approach-to-understanding-nascent-and-mature-mrna/]
- 83. Development of an End-to-End Total RNA Sequencing ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157825001436]
- 84. dtm2451/dittoSeq: Color blindness friendly visualization of ... [https://github.com/dtm2451/dittoSeq]
- 85. spatially aware color palette optimization for single-cell and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9272793/]
- 86. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 87. Integrated analysis of single-cell RNA-seq and bulk RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548165/]
- 88. RnaXtract, a tool for extracting gene expression, variants ... [https://www.nature.com/articles/s41598-025-16875-9]
- 89. Cell type-specific inference from bulk RNA-sequencing data ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03847-5]
- 90. RNA-Seq Analysis - Center for Genetic Medicine [https://www.cgm.northwestern.edu/cores/nuseq/services/bioinformatics/rna-seq-analysis.html]
- 91. Targeted DNA RNA Market Size to Hit USD 70.55 Billion... Sequencing [https://www.precedenceresearch.com/targeted-dna-rna-sequencing-market]
- 92. Searchlight: automated bulk RNA-seq exploration and ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04321-2]
- 93. Cross-modal integration of bulk -seq and single-cell RNA ... RNA [https://pubmed.ncbi.nlm.nih.gov/39344205/]
- 94. RNA-Seq Data Network Analysis [https://cytoscape.org/cytoscape-tutorials/protocols/rna-seq-data-analysis/]
- 95. From bulk, single-cell to spatial RNA sequencing [https://www.nature.com/articles/s41368-021-00146-0]
- 96. Clinical validation of RNA sequencing for Mendelian disorder ... [https://pubmed.ncbi.nlm.nih.gov/40043707/]
- 97. Bulk RNA sequencing for analysis of post COVID-19 condition ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05117-7]
- 98. 50. Glossary [https://www.sc-best-practices.org/glossary.html]
- 99. Best practices for single-cell analysis across modalities [https://pmc.ncbi.nlm.nih.gov/articles/PMC10066026/]
- 100. -seq Bulk and Processing Pipeline – ENCODE RNA Data Standards [https://www.encodeproject.org/data-standards/encode4-bulk-rna/]
- 101. Terminology [https://www.seqcenter.com/terminology/]