Bulk RNA-seq vs Single-Cell RNA-seq: A Comprehensive Guide to Methods, Applications, and Strategic Choice
This article provides a definitive guide for researchers and drug development professionals navigating the choice between bulk and single-cell RNA sequencing.
Bulk RNA-seq vs Single-Cell RNA-seq: A Comprehensive Guide to Methods, Applications, and Strategic Choice
Abstract
This article provides a definitive guide for researchers and drug development professionals navigating the choice between bulk and single-cell RNA sequencing. It covers the foundational principles of each method, detailing their respective workflows from sample preparation to data output. The content explores specific applications across biomedical research, from differential expression analysis to characterizing tumor heterogeneity and discovering novel cell types. It addresses key practical considerations, including cost, experimental complexity, and data analysis challenges, while also highlighting how these technologies are synergistically combined and validated. The goal is to empower scientists with the knowledge to select the optimal transcriptomic approach for their specific research questions and to understand the future trajectory of the field.
Core Concepts: Understanding the Fundamental Principles of Transcriptomics
What is Bulk RNA-seq? Defining the Population-Averaged Transcriptome
Bulk RNA sequencing (Bulk RNA-seq) is a foundational molecular biology technique designed to profile the average gene expression levels in a sample consisting of a pooled population of cells, entire tissue sections, or biopsies [1] [2]. This method provides a global overview of the transcriptome—the complete set of RNA transcripts in a biological sample—by measuring the collective expression of thousands of genes simultaneously [3]. The "bulk" designation distinguishes it from more recent technologies like single-cell RNA sequencing (scRNA-seq), as it analyzes the RNA from thousands to millions of cells all at once, resulting in a population-averaged gene expression profile [4]. This approach has been a cornerstone in transcriptomic studies for nearly two decades, enabling researchers to conduct comparative analyses between different sample conditions, such as healthy versus diseased tissues, or treated versus untreated groups [2] [4]. By capturing the overall transcriptional output of a cell population, bulk RNA-seq serves as a powerful tool for identifying broad gene expression differences, discovering biomarkers, and understanding global regulatory mechanisms [3] [4].
Core Principles and Technical Workflow
The fundamental principle of bulk RNA-seq is the conversion of RNA molecules into complementary DNA (cDNA) libraries that are suitable for high-throughput sequencing on next-generation platforms [3]. A standard bulk RNA-seq workflow involves multiple critical steps, from sample preparation to computational analysis, each contributing to the quality and reliability of the final data.
Sample Preparation and Library Construction
The process begins with RNA extraction from the bulk sample, which could be a cell pellet, a piece of tissue, or a tissue biopsy [1] [5]. Given that ribosomal RNA (rRNA) constitutes over 80% of total RNA but is often not the focus of studies, it is typically removed to enrich for the RNA species of interest [3]. This is achieved through either ribo-depletion (removal of rRNA) or poly(A) selection (enrichment for messenger RNA by capturing its poly-A tail) [3] [5]. The choice between these methods depends on the research goals; poly(A) selection is suitable for studying mature mRNA, while rRNA depletion allows for the analysis of other RNA species, including non-coding RNAs and pre-mRNA [5].
The purified RNA is then converted into a sequenceable library. This involves fragmenting the RNA, reverse transcribing it into double-stranded cDNA, and ligating platform-specific adapters [4] [5]. A critical step in multiplexed studies is the addition of unique molecular barcodes (or indices) to each sample during library preparation. This allows multiple samples to be pooled and sequenced together while maintaining the ability to computationally distinguish them after sequencing [1]. The quality and concentration of the resulting cDNA library are assessed using systems like the Agilent TapeStation before sequencing [1].
Sequencing and Data Analysis
The pooled libraries are sequenced using high-throughput platforms, most commonly from Illumina, generating millions of short reads [4]. The data analysis workflow can be broadly divided into upstream and downstream processes:
Upstream Analysis: This involves quality control of the raw sequencing reads (e.g., using FastP), alignment to a reference genome or transcriptome using splice-aware aligners like STAR, and quantification of gene expression levels [6] [7]. Tools such as Salmon or kallisto use pseudo-alignment to quickly estimate transcript abundances, while others like RSEM employ expectation-maximization algorithms to quantify gene and isoform levels from aligned reads [6] [8]. The final output is a count matrix, where rows represent genes and columns represent samples, containing expression values such as Counts, TPM (Transcripts Per Million), or FPKM (Fragments Per Kilobase of Transcript per Million) [6] [8].
Downstream Analysis: Once the expression matrix is obtained, researchers can perform various data mining operations. These include differential expression analysis to identify genes with significant expression changes between conditions (using tools like DESeq2, EdgeR, or limma) [6] [7], clustering and visualization (e.g., PCA, heatmaps) to explore sample relationships, and enrichment analysis (e.g., GSEA, ORA) to uncover biological pathways or functions associated with the differentially expressed genes [4] [7].
Table 1: Key Steps in a Standard Bulk RNA-seq Workflow
| Stage | Key Steps | Common Tools/Methods |
|---|---|---|
| Sample Prep | RNA Extraction, rRNA depletion/polyA selection, cDNA synthesis, Adapter Ligation | Oligo(dT) beads, Ribosomal depletion kits |
| Sequencing | High-throughput sequencing on a platform | Illumina, Paired-end/Single-end sequencing |
| Upstream Analysis | Quality Control, Read Alignment, Quantification | FastQC, STAR, Salmon, RSEM |
| Downstream Analysis | Differential Expression, Pathway Analysis, Visualization | DESeq2, limma, GSEA, ggplot2 |
The following diagram illustrates the core steps of a standard bulk RNA-seq workflow, from sample to data, including the optional multiplexing step:
Key Applications in Research
Bulk RNA-seq has broad and well-established utilities across biological research and clinical applications. Its ability to provide a quantitative, genome-wide snapshot of gene expression makes it indispensable for:
Differential Gene Expression Studies: This is the most common application, where bulk RNA-seq is used to compare gene expression profiles between different conditions, such as diseased versus healthy tissues, treated versus untreated samples, or different time points in a developmental process [6] [9]. This helps pinpoint genes with significant expression changes that may be drivers of a biological process or disease state [4].
Biomarker Discovery: By analyzing gene expression patterns across large cohorts of samples, researchers can identify specific genes or gene signatures (biomarkers) that are associated with a particular disease, prognosis, or response to treatment [2] [3]. For example, numerous RNA-seq-based signatures have been developed and validated across major tumor types for cancer classification and prognosis prediction [2].
Detection of Gene Fusions and Alternative Splicing: Beyond gene expression levels, bulk RNA-seq can detect various forms of genomic alterations, including gene fusions—which are well-documented as major cancer drivers—as well as alternative splicing events and novel transcripts [2] [4]. Targeted RNA-seq panels, such as FoundationOne Heme, are used in clinical oncology for therapy selection by detecting these alterations [2].
Pathway and Network Analysis: The global expression data from bulk RNA-seq allows for gene set enrichment analysis and the construction of gene regulatory or co-expression networks (e.g., using WGCNA). This helps in understanding the biological pathways and processes that are perturbed in a given condition [4] [7].
Bulk RNA-seq vs. Single-Cell RNA-seq: A Detailed Comparison
The emergence of single-cell RNA sequencing has revolutionized transcriptomics, but bulk RNA-seq remains a widely used and vital technology. The choice between them depends heavily on the research question, as they offer complementary strengths and limitations.
Table 2: Comparing Bulk RNA-seq and Single-Cell RNA-seq
| Aspect | Bulk RNA Sequencing | Single-Cell RNA Sequencing |
|---|---|---|
| Resolution | Population-averaged gene expression [2] [4] | Individual cell resolution [2] [9] |
| Primary Goal | Compare average expression between sample groups [4] | Investigate cellular heterogeneity and identify cell types/states [4] [9] |
| Cost | Relatively low (~1/10th of scRNA-seq) [9] | Higher [9] |
| Data Complexity | Lower, simpler to analyze [9] | High, requires specialized computational methods [9] |
| Cell Heterogeneity | Masks heterogeneity; cannot detect rare cell types [2] [4] | Excellent for uncovering heterogeneity and rare cell populations [2] [9] |
| Gene Detection Sensitivity | Higher per sample, detects more genes [9] | Lower per cell, sparsity due to technical dropout [9] |
| Ideal For | Homogeneous samples, large-scale studies, biomarker discovery, differential expression [4] [9] | Complex tissues, tumor heterogeneity, immune profiling, developmental biology [2] [9] |
The core difference is visualized in the following diagram, which contrasts the averaging nature of bulk RNA-seq with the high-resolution view of scRNA-seq:
Standards, Protocols, and the Scientist's Toolkit
Robust and reproducible bulk RNA-seq experiments rely on adherence to established standards and the use of high-quality reagents.
Experimental and Data Standards
Consortiums like ENCODE have developed rigorous standards for bulk RNA-seq experiments to ensure data quality and reproducibility [8]. Key guidelines include:
- Replication: Experiments should have two or more biological replicates. Replicate concordance is typically measured by Spearman correlation of gene-level quantifications, which should be >0.9 between isogenic replicates [8].
- Sequencing Depth: A common standard is 20-30 million aligned reads per replicate for mRNA-seq libraries, although this can vary based on the experiment's goals [8].
- RNA Quality: RNA Integrity Number (RIN) is a critical metric. A RIN value over 6 is generally considered sufficient for sequencing, though higher is preferable [5].
- Spike-in Controls: Exogenous RNA spike-in controls (e.g., ERCC Spike-ins) are often added to samples during library preparation to provide a standard baseline for quantitative normalization and quality assessment [8].
Successful execution of a bulk RNA-seq project requires a suite of trusted reagents, tools, and software.
Table 3: Essential Toolkit for Bulk RNA-seq Experiments
| Category | Item | Function |
|---|---|---|
| Wet-Lab Reagents | rRNA Depletion Kits / poly-dT Oligos | Enriches for RNA species of interest by removing rRNA or selecting for mRNA [3] [5] |
| Unique Barcoded Adapters | Allows sample multiplexing in a single sequencing run [1] | |
| ERCC Spike-in Controls | Exogenous RNA controls for normalization and QC [8] | |
| Software & Pipelines | STAR | Spliced aligner for mapping reads to a reference genome [6] [8] |
| Salmon / kallisto | Tools for fast, accurate transcript quantification [6] | |
| DESeq2 / limma / EdgeR | Statistical packages for differential expression analysis [6] [7] | |
| Searchlight / nf-core RNA-seq | Automated pipelines for downstream visualization and analysis [7] |
Bulk RNA-seq remains a powerful and indispensable workhorse in the genomic toolbox. Its strength lies in providing a cost-effective, robust, and comprehensive overview of the average transcriptional state of a cell population, making it ideal for differential expression studies, biomarker discovery, and large-scale cohort analyses. While it cannot resolve the cellular heterogeneity captured by single-cell technologies, its well-established protocols, lower cost, and simpler data analysis ensure its continued relevance. The future of transcriptomics lies in integrated approaches, where the broad, quantitative profiling of bulk RNA-seq is combined with the high-resolution insights from scRNA-seq and spatial transcriptomics to build a more complete and multi-scale understanding of complex biological systems [4].
What is Single-Cell RNA-seq? Unveiling Cellular Heterogeneity
Single-cell RNA sequencing (scRNA-seq) represents a transformative technological advancement in genomics that enables researchers to examine the gene expression profile of individual cells. Unlike traditional bulk RNA sequencing, which measures the average gene expression across thousands to millions of cells in a sample, scRNA-seq reveals the cellular heterogeneity and unique transcriptional states of each cell within a complex biological system [9] [10]. Since its conceptual breakthrough in 2009, scRNA-seq has evolved from a specialized technique to an accessible tool that fuels discovery across biomedical research and clinical applications [11] [10]. By uncovering the full diversity of cell types, states, and functions within tissues and organs, scRNA-seq provides an unprecedented window into the fundamental units of biology, revolutionizing our understanding of development, disease, and treatment responses [11] [12].
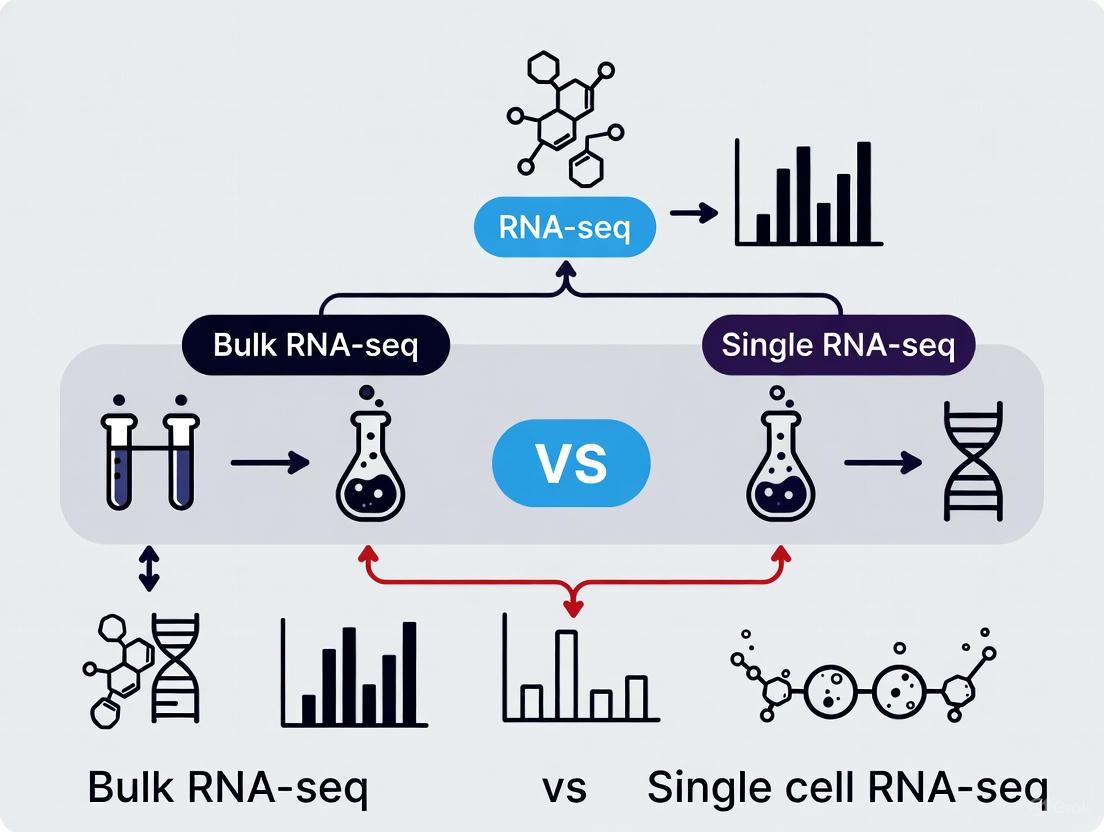
The Fundamental Difference: Bulk RNA-seq vs. Single-Cell RNA-seq
The core distinction between bulk and single-cell RNA sequencing lies in resolution and the resulting biological insights each method can provide.
Bulk RNA-seq processes RNA from a population of cells together, generating a composite gene expression profile that represents the average across all cells in the sample [9] [13]. This approach effectively masks differences between individual cells and obscures rare cell populations.
Single-cell RNA-seq isolates individual cells before RNA capture and sequencing, allowing each cell's transcriptome to be profiled independently [9]. This reveals the cellular composition of tissues, identifies novel cell types, and uncovers continuous transitional states that are invisible in bulk measurements [13] [10].
The following diagram illustrates this fundamental difference in resolution and the type of biological information each method reveals:
The following table provides a detailed quantitative comparison of these two approaches:
Table 1: Key Technical and Practical Differences Between Bulk and Single-Cell RNA-seq
| Feature | Bulk RNA Sequencing | Single-Cell RNA Sequencing |
|---|---|---|
| Resolution | Population average [9] | Individual cell level [9] |
| Cost per Sample | Lower (~$300/sample) [9] | Higher ($500-$2000/sample) [9] |
| Data Complexity | Lower, simpler analysis [9] [14] | Higher, requires specialized computational methods [9] [14] |
| Cell Heterogeneity Detection | Limited, masks diversity [9] [13] | High, reveals cellular diversity [9] [13] |
| Rare Cell Type Detection | Limited or impossible [9] | Possible, can identify rare populations [9] |
| Gene Detection Sensitivity | Higher per sample [9] | Lower per cell, but captures more cell-specific genes [9] |
| Sample Input Requirement | Higher amount of starting material [9] | Lower, can work with minimal cells [9] |
| Ideal Application | Homogeneous samples, differential expression [9] [14] | Complex tissues, rare cell identification, heterogeneity studies [9] [14] |
Technical Framework: How Single-Cell RNA Sequencing Works
The scRNA-seq workflow involves several critical steps to transition from tissue to sequencing data, each with specific technical considerations for preserving cell integrity and transcriptome fidelity.
Single-Cell Isolation and Capture
The initial phase involves creating a viable single-cell suspension from tissue through enzymatic or mechanical dissociation [13]. This step is particularly challenging as the dissociation process can induce artificial transcriptional stress responses, potentially altering the native gene expression profile [11]. Techniques to minimize this include performing dissociations at lower temperatures (4°C) or utilizing single-nucleus RNA sequencing (snRNA-seq) as an alternative, especially for tissues difficult to dissociate, such as brain tissue [11]. Common isolation methods include:
- Fluorescence-Activated Cell Sorting (FACS): Allows selection of specific cell types based on surface markers [11] [10]
- Microfluidic Systems: Enable high-throughput cell capture with minimal hands-on time [11] [15]
- Droplet-Based Partitioning: Currently the most widely used approach, encapsulating individual cells in nanoliter-scale reaction vessels [15] [13]
Library Preparation and Barcoding
After isolation, individual cells are processed to capture and barcode their RNA content. In droplet-based systems like 10x Genomics' platform, single cells are combined with barcoded gel beads and partitioning oil to form GEMs (Gel Beads-in-emulsion) [15] [13]. Within each GEM:
- Cells are lysed to release RNA
- Polyadenylated mRNA molecules hybridize to the poly(T) primers on the gel beads
- Each primer contains a cell barcode (unique to each cell), a unique molecular identifier (UMI), and sequencing adapters [15] [10]
The use of UMIs is critical for accurate transcript quantification as they label individual mRNA molecules, enabling correction for amplification biases during computational analysis [11].
Reverse Transcription, Amplification, and Sequencing
After barcoding, reverse transcription occurs within the partitions to create cDNA libraries where all transcripts from a single cell share the same barcode [15]. The barcoded cDNA is then:
- Amplified via PCR to create sufficient material for sequencing
- Pooled for traditional bulk next-generation sequencing (NGS) [13]
Despite the initial single-cell partitioning, the actual sequencing step is performed as a pooled library, with the cellular origin of each transcript later decoded based on its barcode [15] [10].
The following diagram illustrates this complex workflow from sample preparation to sequencing:
Essential Research Tools: The Single-Cell RNA-seq Toolkit
Successful scRNA-seq experiments require specialized reagents and platforms designed to handle the unique challenges of working with minute quantities of starting material while maintaining cell integrity and transcriptome representation.
Table 2: Key Research Reagent Solutions for Single-Cell RNA-seq
| Reagent/Platform | Function | Technical Considerations |
|---|---|---|
| Cell Barcoding Beads | Gel beads containing barcoded oligonucleotides for labeling cellular origin [15] | Critical for sample multiplexing and cell identity preservation |
| Unique Molecular Identifiers (UMIs) | Random nucleotide sequences that uniquely tag individual mRNA molecules [11] | Enables accurate transcript quantification by correcting PCR amplification bias |
| Reverse Transcription Mix | Converts mRNA to cDNA with template-switching capability [11] [10] | SMARTer chemistry improves full-length transcript capture |
| Partitioning Oil & Microfluidics | Creates stable water-in-oil emulsions for single-cell isolation [15] [13] | GEM-X technology increases throughput while reducing multiplet rates |
| Library Preparation Kits | Prepares barcoded cDNA for next-generation sequencing [15] [10] | Compatibility with various sequencing platforms (Illumina, PacBio, Oxford Nanopore) |
| Cell Viability Stains | Assesses cell integrity and membrane integrity before processing [13] | Critical for ensuring high-quality input material and reducing ambient RNA |
| Enzymatic Dissociation Kits | Tissue-specific protocols for generating single-cell suspensions [13] | Must balance yield with preservation of native transcriptional states |
Commercial scRNA-seq platforms such as 10x Genomics' Chromium system, Parse Biosciences' Evercode combinatorial barcoding, and Fluidigm's C1 platform provide integrated solutions that combine many of these reagent systems into standardized workflows [15] [16]. The choice between platforms depends on specific research needs, including cell throughput, sequencing depth, cost constraints, and sample type (fresh, frozen, or fixed) [15] [13].
Applications in Drug Discovery and Development
The pharmaceutical industry has embraced scRNA-seq as a powerful tool to address key challenges in drug discovery and development, particularly in understanding disease mechanisms, identifying therapeutic targets, and predicting clinical responses.
Target Identification and Validation
scRNA-seq enables cell-type-specific target discovery by identifying genes expressed specifically in disease-relevant cell populations [12] [16]. For example, in a comprehensive analysis across 30 diseases and 13 tissues, researchers found that drug targets with cell-type-specific expression in disease-relevant tissues were more likely to progress successfully from Phase I to Phase II clinical trials [16]. This predictive power helps prioritize targets with higher potential for success early in the development process.
When combined with CRISPR-based functional genomics, scRNA-seq can simultaneously measure the transcriptomic effects of thousands of genetic perturbations in individual cells [12] [16]. This approach maps regulatory networks and pathway dependencies, providing mechanistic insights into how potential targets contribute to disease pathology [16].
Biomarker Discovery and Patient Stratification
The technology has proven particularly valuable in oncology, where it reveals intratumoral heterogeneity and identifies resistance mechanisms to therapy [9] [12]. For instance, scRNA-seq studies of melanoma patients receiving checkpoint inhibitor immunotherapy identified distinct T cell states associated with treatment response or resistance [12]. These findings enable development of predictive biomarkers for patient selection and treatment monitoring.
Similar approaches have identified rare cell populations driving disease progression in multiple myeloma and non-small cell lung cancer, revealing potential biomarkers for disease monitoring and novel therapeutic targets [9] [12].
Toxicity Assessment and Mechanism of Action Studies
By profiling drug responses across diverse cell types within complex tissues, scRNA-seq provides comprehensive safety assessments early in drug development [17] [16]. This approach can identify cell-type-specific toxicities that might be missed in traditional bulk analyses, potentially reducing late-stage attrition due to safety concerns [16].
Furthermore, scRNA-seq elucidates drug mechanisms of action by capturing how different cell populations within a tissue respond to treatment, informing both efficacy and safety profiles [12]. This detailed understanding supports more rational drug design and optimization.
Single-cell RNA sequencing has fundamentally transformed our ability to study biology and disease at its most basic resolution—the individual cell. By revealing the cellular heterogeneity that underlies tissue function, developmental processes, and disease mechanisms, scRNA-seq provides insights that were previously inaccessible with bulk sequencing approaches. As technologies continue to evolve with decreasing costs, increased throughput, and enhanced multimodal capabilities (combining transcriptomics with epigenomics, proteomics, and spatial information), scRNA-seq is poised to become an increasingly central tool in both basic research and translational applications [9] [11] [12]. For drug discovery professionals and researchers, mastering this technology and its applications offers the potential to accelerate the development of more targeted, effective, and personalized therapeutic interventions.
In the study of gene expression, two powerful technologies offer fundamentally different vantage points. Bulk RNA sequencing (bulk RNA-seq) provides a population-level overview, akin to observing a forest from a distance, while single-cell RNA sequencing (scRNA-seq) reveals the individual trees, capturing the unique characteristics of each cell [13]. This distinction is critical for researchers, scientists, and drug development professionals navigating the complexities of biological systems and disease mechanisms. The choice between these methods—or the decision to integrate them—carries significant implications for experimental design, cost, analytical capabilities, and ultimately, the biological insights that can be gained [13] [18]. This technical guide explores the core differences, applications, and methodologies of these two approaches, providing a structured framework for their application in modern research.
Core Concepts and Definitions
Bulk RNA Sequencing: The Population Perspective
Bulk RNA-seq is a next-generation sequencing (NGS)-based method designed to measure the whole transcriptome across a population of thousands to millions of cells [13]. It operates as a whole population approach, processing a biological sample where many different cells are pooled together. The resulting data represents an average readout of gene expression levels for individual genes across all cells within the sample [13]. The foundational workflow involves digesting the biological sample to extract RNA, which is then converted into cDNA before library preparation and sequencing [13]. Its key strength lies in providing a holistic, cost-effective view of the average gene expression profile of an entire tissue sample or cell population [19].
Single-Cell RNA Sequencing: The Cellular Perspective
Single-cell RNA sequencing (scRNA-seq) represents a paradigm shift, enabling the measurement of whole transcriptome gene expression profiles for each individual cell within a sample [13]. This cellular-resolution approach fundamentally changes how scientists observe biological systems, transforming a homogenous average into a detailed landscape of cellular heterogeneity. The scRNA-seq workflow necessitates several specialized steps, beginning with the generation of viable single cell suspensions from whole samples through enzymatic or mechanical dissociation [13] [20]. A critical differentiator from bulk methods is the instrument-enabled cell partitioning, where single cells are isolated into individual micro-reaction vessels—such as the Gel Beads-in-emulsion (GEMs) used in 10x Genomics' Chromium system—ensuring that analytes from each cell can be traced back to their origin through cell-specific barcodes [13] [20].
Table 1: Fundamental Comparison of Bulk RNA-seq and Single-Cell RNA-seq
| Feature | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Population-level average [13] | Individual cell level [13] |
| Primary Output | Averaged gene expression across all cells [13] | Cell-to-cell variation and heterogeneity [13] |
| Key Advantage | Cost-effective for large studies; established analysis pipelines [19] | Reveals rare cell types, novel cell states, and cellular dynamics [13] |
| Sample Input | Pooled cells from tissue or population [13] | Viable single-cell suspension [13] |
| Typical Cost | Lower cost per sample [13] [19] | Higher cost per sample, but decreasing [13] |
| Data Complexity | Lower; more straightforward analysis [13] | High-dimensional; requires specialized bioinformatics [13] [21] |
| Ideal Use Case | Differential expression between conditions; biomarker discovery [13] | Characterizing heterogeneous populations; developmental trajectories [13] |
Technical and Experimental Comparisons
Divergent Experimental Workflows
The experimental protocols for bulk and single-cell RNA-seq diverge significantly from the initial sample preparation stage, each presenting unique technical considerations and challenges.
Bulk RNA-seq Protocol follows a relatively straightforward path [13]:
- Sample Digestion: Biological sample is digested to extract total RNA or enriched mRNA.
- cDNA Conversion: RNA is converted into cDNA.
- Library Preparation: Processing steps prepare a sequencing-ready gene expression library.
- Sequencing & Analysis: Sequencing is performed followed by data analysis to review gene expression levels across the tissue sample.
Single-Cell RNA-seq Protocol requires more specialized steps to preserve single-cell resolution [13] [20]:
- Sample Dissociation: Generation of viable single-cell suspensions via enzymatic or mechanical dissociation.
- Quality Control: Cell counting and viability assessment to ensure sample quality.
- Cell Partitioning: Single cells are isolated into individual micro-reaction vessels using microfluidic systems.
- Cell Lysis & Barcoding: Cells are lysed within partitions, and RNA is captured and barcoded with cell-specific barcodes.
- Library Preparation & Sequencing: Barcoded products are used to create sequencing libraries for whole transcriptome measurement.
Analytical Approaches and Computational Considerations
The data analysis pipelines for bulk and single-cell RNA-seq differ substantially in complexity and methodology, reflecting their fundamentally different data structures and research questions.
Bulk RNA-seq Analysis typically involves established, relatively straightforward pipelines [19]:
- Differential gene expression analysis using tools like DESeq2 or edgeR
- Pathway and enrichment analysis (GO, KEGG)
- Biomarker identification and validation
Single-Cell RNA-seq Analysis requires specialized computational approaches to handle high-dimensional data [21] [20] [22]:
- Quality Control & Preprocessing: Filtering cells based on detected genes, mitochondrial content, and other QC metrics
- Dimensionality Reduction: Principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP)
- Clustering & Cell Type Identification: Graph-based clustering algorithms and marker gene detection for cell type annotation
- Trajectory Inference: Reconstruction of developmental pathways using tools like Monocle
- Cell-Cell Communication Analysis: Inference of ligand-receptor interactions using tools like CellPhoneDB
Machine learning has become increasingly integral to scRNA-seq analysis, with applications in clustering, dimensionality reduction, trajectory inference, and cell type annotation [22]. Advanced computational frameworks like SEURAT and Galaxy Europe Single Cell Lab provide essential bioinformatic tools for analyzing these complex datasets [20].
Applications and Synergistic Integration
Distinct and Complementary Applications
Each technology excels in addressing specific biological questions, with their applications reflecting their fundamental resolutions.
Key Applications of Bulk RNA-seq [13] [19]:
- Differential Gene Expression Analysis: Comparing gene expression profiles between different experimental conditions (disease vs. healthy, treated vs. control)
- Biomarker Discovery: Identifying molecular signatures for diagnosis, prognosis, or disease stratification
- Pathway Analysis: Investigating how sets of genes change collectively under various biological conditions
- Tissue-Level Transcriptomics: Obtaining global expression profiles from whole tissues, organs, or bulk-sorted cell populations
- Large Cohort Studies: Profiling large sample sets in biobank projects or population-level studies
Key Applications of Single-Cell RNA-seq [13] [20]:
- Characterizing Heterogeneous Populations: Identifying novel cell types, cell states, and rare cell populations
- Developmental Lineage Reconstruction: Tracing cellular differentiation pathways and developmental hierarchies
- Tumor Microenvironment Analysis: Deconvoluting complex tissue ecosystems and cell-cell interactions
- Cell-Type Specific Responses: Understanding how individual cells respond to stimuli, disease, or treatment
- Identifying Rare Driver Populations: Discovering rare cell types or transient states that play key roles in disease biology
Table 2: Application-Based Selection Guide
| Research Goal | Recommended Approach | Rationale |
|---|---|---|
| Differential Expressionbetween conditions | Bulk RNA-seq [13] | Cost-effective for large sample numbers; statistical power for population-level differences |
| Novel Cell Type Discovery | Single-Cell RNA-seq [13] | Unbiased identification of previously unrecognized subpopulations |
| Rare Cell Population Detection(e.g., circulating tumor cells) | Single-Cell RNA-seq [18] | Sensitivity to identify ultra-rare populations that bulk methods would miss |
| Large Cohort Studies(e.g., clinical trials) | Bulk RNA-seq [13] [19] | Practical for processing hundreds to thousands of samples cost-effectively |
| Developmental Trajectories(e.g., embryogenesis) | Single-Cell RNA-seq [13] [18] | Reconstruction of differentiation pathways and lineage relationships |
| Pathway Analysisacross tissues | Bulk RNA-seq [13] | Comprehensive snapshot of average pathway activity across entire tissue |
| Cell-Cell Communicationin tissues | Single-Cell RNA-seq [23] [24] | Identification of ligand-receptor interactions between specific cell types |
| Therapeutic Target Identification | Integrated Approach [18] [24] | Bulk identifies candidate pathways; single-cell pinpoints specific cellular targets |
Synergistic Integration in Research
Rather than competing technologies, bulk and single-cell RNA-seq increasingly function as complementary tools that provide deeper insights when integrated [18] [24]. This synergy enables researchers to navigate efficiently between population-level trends and cellular-resolution mechanisms.
Successful Integration Framework [18]:
- Hypothesis Generation: Use bulk data to identify pathways of interest across entire tissues or conditions
- Cellular Resolution: Apply single-cell sequencing to pinpoint which specific cells drive the observed changes
- Validation & Scaling: Confirm single-cell discoveries across larger patient cohorts or timepoints with bulk sequencing
- Multi-omics Integration: Combine genomic, transcriptomic, and epigenomic layers for a systems-level view
A compelling example of this synergy comes from Huang et al. (2024), who leveraged both bulk and single-cell RNA-seq in healthy human B cells and leukemia clinical samples to identify developmental states driving resistance and sensitivity to asparaginase chemotherapy in B-cell acute lymphoblastic leukemia (B-ALL) [13]. Similarly, research on rheumatoid arthritis integrated scRNA-seq and bulk RNA-seq to elucidate macrophage heterogeneity, identifying STAT1 as a key regulator in disease pathogenesis through combined analytical approaches [24].
The Scientist's Toolkit: Essential Research Solutions
Successful implementation of RNA sequencing technologies requires familiarity with key platforms, reagents, and computational resources that constitute the modern transcriptomics toolkit.
Table 3: Research Reagent Solutions and Essential Materials
| Tool Category | Specific Examples | Function & Application |
|---|---|---|
| Single-Cell Platforms | 10x Genomics Chromium System [13]Fluidigm C1 SystemPlate-based FACS [20] | Instrument-enabled cell partitioning and barcoding for high-throughput scRNA-seq |
| Library Prep Kits | 10x Genomics GEM-X Flex [13]10x Genomics Universal Multiplex Assays [13] | Preparation of sequencing-ready libraries with cell barcoding and UMIs |
| Bioinformatics Tools | Seurat [21] [23] [24]Monocle [23]CellPhoneDB [23] [24] | Analysis pipelines for scRNA-seq data including clustering, trajectory inference, and cell-cell communication |
| Quality Control Tools | Cell Ranger [21]DoubletFinder [24]InferCNV [23] | QC metrics, doublet detection, and copy number variation analysis for single-cell data |
| Data Integration Tools | Harmony Algorithm [24]CIBERSORT [23] | Batch effect correction and cell type deconvolution in single-cell and bulk data |
| Visualization Software | Loupe Browser (10x Genomics) [20]UCSC Cell Browser | Interactive visualization and exploration of single-cell datasets |
Signaling Pathway Case Study: STAT1 in Rheumatoid Arthritis
Recent research on rheumatoid arthritis (RA) provides an illustrative example of how integrated bulk and single-cell approaches can elucidate key signaling pathways in disease pathogenesis. This case study exemplifies the powerful synergy between these technologies.
Experimental Findings [24]:
- scRNA-seq Analysis: Revealed expanded population of Stat1+ macrophages in RA synovial tissue with inflammatory pathway enrichment
- Bulk RNA-seq Validation: Confirmed STAT1 upregulation across multiple RA patient cohorts (GSE12021, GSE55235, GSE55457, GSE77298, GSE89408)
- Functional Experiments: Demonstrated that STAT1 activation upregulates LC3 and ACSL4 while downregulating p62 and GPX4, suggesting modulation of autophagy and ferroptosis pathways
- Therapeutic Intervention: Fludarabine treatment reversed these changes, positioning STAT1 as a potential therapeutic target
This pathway analysis exemplifies how scRNA-seq identified a specific cellular subpopulation (Stat1+ macrophages) driving RA pathology, while bulk RNA-seq provided statistical validation across cohorts, together revealing a mechanistically coherent pathway with therapeutic implications.
The evolution of transcriptomics continues to advance toward higher resolution and integration. While bulk RNA-seq remains invaluable for population-level studies and large-scale screening, single-cell RNA-seq has fundamentally transformed our understanding of cellular heterogeneity and complexity [20] [25]. The future lies not in choosing between these approaches, but in strategically integrating them to leverage their complementary strengths [18].
Emerging technologies, particularly spatial transcriptomics, are now bridging the gap between single-cell resolution and tissue context, adding another dimension to the transcriptomics toolkit [25]. Meanwhile, advances in machine learning and artificial intelligence are addressing computational challenges in scRNA-seq data analysis, enabling more sophisticated integration of multi-omics datasets and accelerating the translation of single-cell discoveries into clinical applications [22].
For researchers and drug development professionals, this evolving landscape offers unprecedented opportunities to understand biological systems and disease mechanisms. By strategically employing both the "forest" perspective of bulk RNA-seq and the "tree" perspective of single-cell RNA-seq—and increasingly, the "geographic map" of spatial transcriptomics—we can continue to unravel the complexity of biological systems and advance the development of targeted therapeutics and precision medicine.
In the field of genomics, two principal methodologies have emerged for profiling gene expression: bulk RNA sequencing (bulk RNA-seq) and single-cell RNA sequencing (scRNA-seq). While both techniques utilize next-generation sequencing to capture a snapshot of the transcriptome, they differ fundamentally in their resolution and application. Bulk RNA-seq provides a population-level average of gene expression by sequencing RNA extracted from thousands to millions of cells simultaneously [13] [26]. In contrast, scRNA-seq enables researchers to measure gene expression at the resolution of individual cells, revealing the cellular heterogeneity masked in bulk measurements [13] [27]. This technical guide provides a comprehensive, step-by-step comparison of the experimental workflows for both approaches, detailing procedures from sample preparation through sequencing to empower researchers in selecting and implementing the appropriate method for their biological questions.
Bulk RNA-Seq Experimental Workflow
Sample Preparation and RNA Extraction
The bulk RNA-seq workflow begins with the collection of tissue or cell samples. Unlike single-cell methods, bulk protocols start with a population of cells that are processed together. RNA is extracted from the entire sample using methods that preserve RNA integrity, with careful consideration given to RNA quality and quantity [13]. For standard mRNA sequencing, extraction typically yields total RNA, which may undergo subsequent enrichment steps. The required sample input is substantially higher than for single-cell methods, as sufficient RNA must be obtained to represent the entire cell population [28].
Library Preparation: Whole Transcriptome vs. 3' mRNA-Seq
A critical distinction in bulk RNA-seq library preparation lies in the choice between whole transcriptome and 3' mRNA-seq approaches [28]:
Whole Transcriptome Sequencing: This approach utilizes random primers during cDNA synthesis, resulting in sequencing reads distributed across the entire length of transcripts. It requires either enrichment for polyadenylated RNA (poly(A) selection) or depletion of ribosomal RNA (rRNA) to prevent unnecessary sequencing of highly abundant ribosomal RNA. Whole transcriptome sequencing provides comprehensive information including alternative splicing, novel isoforms, fusion genes, and non-coding RNAs, but requires longer workflow times and deeper sequencing [28].
3' mRNA-Seq: This method focuses sequencing on the 3' end of transcripts through an initial oligo(dT) priming step, effectively performing in-preparation poly(A) selection. By generating one fragment per transcript, 3' mRNA-seq simplifies both library preparation and downstream data analysis. It is ideal for accurate gene expression quantification, particularly for large-scale studies or when working with challenging sample types like FFPE or degraded RNA [28].
Following RNA extraction and optional enrichment, the general workflow proceeds through cDNA synthesis, adapter ligation, and PCR amplification to create sequencing-ready libraries [13].
Sequencing and Data Analysis
Bulk RNA-seq libraries are typically sequenced to a depth sufficient for the research question, with differential gene expression studies often requiring 20-50 million reads per sample [28]. Following sequencing, data analysis involves quality control, read alignment to a reference genome, and gene expression quantification. Differential expression analysis between conditions then identifies genes that are statistically significantly upregulated or downregulated [13].
Table 1: Key Applications and Technical Requirements for Bulk RNA-seq
| Parameter | Whole Transcriptome Approach | 3' mRNA-Seq Approach |
|---|---|---|
| Primary Application | Alternative splicing, isoform discovery, fusion genes, non-coding RNAs [28] | Gene expression quantification, large-scale studies [28] |
| Library Prep Complexity | Higher (requires rRNA depletion or polyA selection) [28] | Lower (streamlined with oligo(dT) priming) [28] |
| Sequencing Depth | Higher (reads distributed across transcripts) [28] | Lower (1-5 million reads/sample often sufficient) [28] |
| Ideal Sample Types | High-quality RNA, discovery projects [28] | Degraded RNA, FFPE, high-throughput screens [28] |
| Data Analysis Complexity | Higher (splice-aware alignment, isoform resolution) [28] | Lower (read counting for expression) [28] |
Single-Cell RNA-Seq Experimental Workflow
Generation of Single-Cell Suspensions
The most critical and technically challenging step in scRNA-seq is the creation of high-quality single-cell suspensions [13]. This process varies significantly based on the starting material (e.g., fresh tissue, frozen tissue, or blood) and requires optimization to maximize cell viability while preserving transcriptional states. Tissue dissociation typically involves enzymatic or mechanical methods, or a combination of both [13]. Strict quality control is essential at this stage, with cell counting and viability assessment performed to ensure samples contain an appropriate concentration of viable cells and are free of clumps and debris [13]. Poor cell viability can lead to elevated ambient RNA, which compromises data quality.
Single-Cell Partitioning and Barcoding
Unlike bulk RNA-seq, scRNA-seq requires physical separation of individual cells before library preparation. The 10x Genomics Chromium system, a widely used platform, achieves this through microfluidic partitioning, where single cells are isolated into nanoliter-scale reaction vessels called Gel Beads-in-emulsion (GEMs) [13]. Within each GEM, gel beads dissolve to release oligonucleotides containing unique cellular barcodes that label all transcripts from an individual cell. Cell lysis occurs within the partition, allowing released RNA to be captured and barcoded [13]. This critical step ensures that all sequencing reads can be traced back to their cell of origin during data analysis. Alternative methods for single-cell isolation include plate-based approaches with FACS sorting [29].
Library Preparation and Sequencing
Following barcoding, the products from all GEMs are pooled, and cDNA is synthesized and amplified. Sequencing libraries are then constructed to be compatible with next-generation sequencing platforms [13]. scRNA-seq experiments typically require significantly deeper sequencing than bulk experiments, as each cell must be sequenced adequately to detect a sufficient number of genes. Current high-throughput scRNA-seq methods can profile thousands to tens of thousands of cells in a single experiment, generating enormous datasets that require specialized computational tools for analysis [13] [26].
Special Considerations: Whole Transcriptome vs. Targeted scRNA-seq
A key methodological choice in single-cell research is between whole transcriptome and targeted gene expression profiling [29]:
Whole Transcriptome scRNA-seq: This unbiased approach aims to capture the entire transcriptome of each cell, making it ideal for discovery-oriented applications such as novel cell type identification, constructing cellular atlases, and uncovering novel disease pathways. However, it spreads sequencing reads thinly across all ~20,000 genes, making it susceptible to "gene dropout" where low-abundance transcripts fail to be detected [29].
Targeted scRNA-seq: This approach focuses sequencing resources on a predefined panel of tens to hundreds of genes. By concentrating reads on genes of interest, it achieves superior sensitivity and quantitative accuracy for those targets, minimizes gene dropout, and is more cost-effective for large-scale studies. The major limitation is its inability to detect genes outside the predefined panel [29].
Table 2: Comparison of Single-Cell RNA-seq Methodologies
| Parameter | Whole Transcriptome scRNA-seq | Targeted scRNA-seq |
|---|---|---|
| Primary Application | Discovery research, novel cell type identification, atlas building [29] | Targeted hypothesis testing, biomarker validation, large cohorts [29] |
| Sensitivity for Low-Abundance Transcripts | Lower (spreads reads across all genes) [29] | Higher (concentrates reads on gene panel) [29] |
| Cost Per Cell | Higher [29] | Lower [29] |
| Scalability for Large Cohorts | Lower cost and computational burden [29] | Higher (enables hundreds/thousands of samples) [29] |
| Computational Complexity | Higher (high-dimensional data) [29] | Lower (focused data analysis) [29] |
Comparative Workflow Visualization
The following diagram illustrates the key procedural differences between bulk and single-cell RNA-seq workflows from sample to sequence:
Experimental Design Considerations
Sample Size and Replication
Appropriate experimental design is crucial for generating meaningful transcriptomic data. For bulk RNA-seq, recent large-scale empirical studies in mouse models suggest that sample sizes of 6-7 represent a minimum, with 8-12 replicates per condition providing significantly more reliable results [30]. Studies with only 3-4 replicates show high false positive rates and fail to detect many truly differentially expressed genes [30]. For scRNA-seq, the concept of replication operates at two levels: the number of biological replicates (individuals) and the number of cells sequenced per sample. While there are no universal standards for scRNA-seq sample sizes, larger numbers of individuals provide greater biological generality, while sequencing more cells enables better characterization of rare cell populations [31].
Method Selection Guide
The choice between bulk and single-cell RNA-seq depends primarily on the research question:
Choose Bulk RNA-seq when:
- The research goal is to identify average expression changes across entire tissues or cell populations [13]
- Studying homogeneous cell populations where cellular heterogeneity is not a primary concern [26]
- Resource constraints exist (lower cost per sample) [13] [27]
- Analyzing large clinical cohorts or time-series studies [27]
Choose scRNA-seq when:
- Characterizing cellular heterogeneity, identifying rare cell types, or discovering novel cell states [13] [26]
- Investigating complex tissues with diverse cell types (e.g., tumors, brain, developing tissues) [13]
- Tracing developmental trajectories or lineage relationships [13]
- Understanding cell-specific responses to perturbations or disease [13]
Complementary Approaches
Bulk and single-cell RNA-seq are not mutually exclusive; they can be powerfully combined in the same research program [13] [23]. For example, bulk RNA-seq can identify global expression changes between conditions, while follow-up scRNA-seq can determine which specific cell types drive these changes [13]. Similarly, discoveries from exploratory scRNA-seq studies (e.g., novel cell types or biomarkers) can be validated across large patient cohorts using targeted scRNA-seq or bulk RNA-seq [29]. A 2024 study on B-cell acute lymphoblastic leukemia exemplified this integrated approach, using both technologies to identify distinct cellular states driving chemotherapy resistance [13].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for RNA-seq Workflows
| Reagent/Material | Function | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|---|
| RNA Stabilization Reagents | Preserve RNA integrity post-collection [32] | Essential | Critical (due to longer processing) |
| Cell Dissociation Kits | Tissue dissociation into single cells [13] | Not required | Essential (enzyme/mechanical) |
| Viability Stains | Assess cell viability pre-processing [13] | Optional | Critical (e.g., trypan blue) |
| Poly(dT) Magnetic Beads | mRNA enrichment via polyA tail [28] | Required for 3' mRNA-seq | Incorporated in barcoded beads |
| rRNA Depletion Kits | Remove abundant ribosomal RNA [28] | Required for whole transcriptome | Not typically used |
| Barcoded Gel Beads | Label transcripts with cell barcodes [13] | Not required | Essential (e.g., 10x Genomics) |
| Partitioning Chips/Instrument | Isolate single cells in emulsions [13] | Not required | Essential (e.g., Chromium X) |
| Library Preparation Kits | Prepare sequencing-ready libraries [28] | Multiple options available | Platform-specific kits |
| Unique Molecular Identifiers | Correct for PCR amplification bias [13] | Optional | Essential for quantification |
Bulk and single-cell RNA sequencing offer complementary approaches for studying gene expression, each with distinct workflow requirements and applications. Bulk RNA-seq provides a cost-effective method for identifying population-level expression changes, with choices between whole transcriptome and 3' mRNA-seq approaches impacting the biological information obtained. Single-cell RNA-seq reveals cellular heterogeneity through more complex workflows requiring single-cell suspension, barcoding, and partitioning, with options for whole transcriptome or targeted profiling. The decision between these technologies should be guided by the specific research question, resources, and desired resolution. As both technologies continue to evolve, their integrated application promises to further unravel the complexity of biological systems.
In the field of transcriptomics, bulk and single-cell RNA sequencing (RNA-seq) represent complementary paradigms for quantifying gene expression. Bulk RNA-seq provides a population-average readout, measuring the composite gene expression profile from a mixture of cells [13] [9]. In contrast, single-cell RNA-seq (scRNA-seq) resolves expression to the level of individual cells, capturing the cell-to-cell variation that is averaged out in bulk approaches [33] [2]. This technical guide explores the interpretive frameworks for these key outputs, enabling researchers to select the appropriate tool for their biological questions, particularly in drug discovery and development.
Fundamental Technical Differences and Output Interpretations
The core difference between these methodologies lies not just in protocol, but in the fundamental nature of the data they generate and the biological conclusions they support.
Bulk RNA-seq: The Population Average
- The "Forest" View: Bulk RNA-seq treats a tissue or cell population as a single entity [13]. The final output is an average expression level for each gene across all cells in the sample.
- Interpretation of Outputs: Results represent the global transcriptional state of the sample. A highly expressed gene in bulk data could mean it is moderately expressed in all cells or very highly expressed in a rare subpopulation. This ambiguity is its primary limitation [9] [2].
- Ideal Use Cases: Identifying large, consistent expression changes between conditions (e.g., diseased vs. healthy tissue), transcriptome annotation, and detecting alternative splicing or gene fusions [13] [34].
Single-Cell RNA-seq: The Cellular Resolution
- The "Individual Tree" View: scRNA-seq profiles the transcriptome of each cell individually [13]. The key output is a cell-by-gene expression matrix, where each value represents the expression level of a specific gene in a specific cell.
- Interpretation of Outputs: This matrix enables the dissection of cellular heterogeneity. Researchers can identify distinct cell types and states, trace developmental lineages, and discover rare cell populations that would be statistically or biologically masked in bulk data [33] [9].
- Ideal Use Cases: Characterizing complex tissues (e.g., tumor microenvironments, immune cells), identifying novel cell types, studying developmental trajectories, and understanding drug resistance mechanisms [35] [2].
Table 1: Core Methodological and Output Comparison of Bulk vs. Single-Cell RNA-seq
| Feature | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Population average [13] [9] | Individual cell level [13] [9] |
| Key Output | Average gene expression profile for the sample [13] | Cell-by-gene expression matrix [13] |
| Heterogeneity Insight | Masks cellular heterogeneity [9] [2] | Reveals cellular heterogeneity, rare cell types, and continuous states [9] [2] |
| Cost per Sample | Lower [9] | Significantly higher [9] |
| Data Complexity | Lower, more established analytical pipelines [9] | Higher, requires specialized tools for sparse data [35] [9] |
| Gene Detection Sensitivity | Higher per sample, detects more lowly expressed genes [9] | Lower per cell due to dropout events, but deeper per cell type [9] |
| Sample Input | Higher RNA input requirements [9] | Can work with very low input, down to single cells [9] |
Experimental Protocols and Workflows
The journey from biological sample to interpretable data involves distinct experimental protocols for bulk and single-cell RNA-seq.
Bulk RNA-seq Workflow
The bulk RNA-seq protocol is a well-established and relatively straightforward process:
- Sample Lysis and RNA Extraction: The entire tissue or cell population is lysed, and total RNA is extracted [13].
- RNA Selection: Either mRNA is enriched using poly-A selection, or ribosomal RNA is depleted to access other RNA species [13].
- Library Preparation: The RNA is reverse-transcribed into cDNA, adapters are ligated, and the library is amplified to create a sequencing-ready pool representing the entire sample's transcriptome [13] [2].
- Sequencing and Analysis: The library is sequenced on an NGS platform, and reads are aligned to a reference genome to quantify gene expression levels for the sample as a whole [13].
Single-Cell RNA-seq Workflow
The scRNA-seq workflow introduces critical steps to preserve single-cell resolution:
- Single-Cell Suspension: The tissue is dissociated into a viable suspension of single cells, requiring careful enzymatic or mechanical processing [13] [2].
- Cell Partitioning and Barcoding: Individual cells are isolated into nanoliter-scale reactions, typically droplets (GEMs in the 10x Genomics platform) or microwells. This is a critical step where each cell's RNA is tagged with a unique cell barcode that distinguishes its origin from all other cells. Unique Molecular Identifiers (UMIs) are also added to correct for PCR amplification bias [13] [2].
- Library Preparation and Sequencing: As in bulk RNA-seq, barcoded cDNA is amplified and prepared for sequencing. The resulting library contains transcriptome data from thousands of cells, multiplexed together [13].
- Bioinformatic Analysis: A specialized computational pipeline (e.g., Cell Ranger) first uses the cell barcodes to demultiplex the data, grouping all reads by their cell of origin to reconstruct the single-cell expression matrix. Subsequent analysis involves quality control, clustering, and cell type annotation [13] [35].
Applications in Drug Discovery and Development
The choice between bulk and single-cell approaches has profound implications for the efficiency and success of drug development pipelines.
Table 2: Application-Specific Advantages in Drug Discovery
| Drug Discovery Stage | Bulk RNA-seq Application | Single-Cell RNA-seq Application |
|---|---|---|
| Target Identification | Differential expression analysis between diseased and healthy bulk tissues to find consistently altered pathways [13] [35]. | Identification of novel cell types or cell states that drive disease within a complex tissue. Discovery of cell-type-specific disease genes [35] [16]. |
| Target Validation | Functional studies in cell lines or animal models using bulk readouts of pathway activity [35]. | High-plex CRISPR screens coupled with scRNA-seq (Perturb-seq) to map gene regulatory networks and understand the function of target genes at single-cell resolution [35]. |
| Biomarker Discovery | Identification of mRNA expression signatures from bulk tissue that correlate with disease status or progression [13] [2]. | Definition of more accurate, cell-type-specific biomarkers. Precise patient stratification based on the abundance or state of specific cell subpopulations (e.g., T-cell states) [35] [16]. |
| Mechanism of Action | Profiling overall transcriptomic changes in response to drug treatment in cell populations [35]. | Uncovering heterogeneous responses to therapy, identifying rare, drug-resistant subpopulations, and understanding how different cells in a tumor microenvironment respond [35] [2]. |
| Toxicity & Safety | Assessing gross transcriptional changes in organs indicative of toxicity [16]. | Pinpointing the specific cell types that are most susceptible to drug-induced toxicity [16]. |
Case Study: Integrating Bulk and Single-Cell Data for Predictive Power
A powerful emerging trend is the integration of bulk and single-cell data to leverage their respective strengths. For instance, a 2024 retrospective analysis of known drug target genes found that cell type-specific expression of a target in disease-relevant tissues, a discovery enabled by scRNA-seq, was a robust predictor of the target's success in progressing from Phase I to Phase II clinical trials [16]. This demonstrates how single-cell resolution can deconvolute the bulk expression signals to provide more precise and predictive insights.
Furthermore, computational frameworks like scDEAL (single-cell Drug rEsponse AnaLysis) use deep transfer learning to predict single-cell drug responses by integrating knowledge from large-scale bulk RNA-seq databases of cancer cell lines (e.g., GDSC, CCLE). This approach harmonizes bulk and single-cell data, transferring trustworthy gene-drug relations from the bulk level to predict response and resistance mechanisms at the single-cell level [36].
The Scientist's Toolkit: Essential Reagents and Platforms
Successful implementation of RNA-seq technologies relies on a suite of specialized reagents and instruments.
Table 3: Key Research Reagent Solutions and Platforms
| Item / Platform | Function / Application |
|---|---|
| 10x Genomics Chromium | An integrated microfluidic system (e.g., Chromium X) for partitioning thousands of single cells into Gel Beads-in-emulsion (GEMs) for 3' or 5' gene expression library prep [13] [2]. |
| Parse Biosciences Evercode | A combinatorial barcoding method that performs scRNA-seq without specialized instruments, enabling very high-throughput experiments (e.g., millions of cells across thousands of samples) [16]. |
| Gel Beads & Barcoded Oligos | Microbeads coated with millions of oligonucleotides containing cell barcodes, UMIs, and poly-dT primers. Critical for labeling all mRNA from a single cell with the same barcode during reverse transcription within a GEM [13] [2]. |
| Enzymatic Mixes (RT, PCR) | Specialized reverse transcriptase and DNA polymerase mixes optimized for the unique challenges of single-cell workflows, including working with very low starting RNA quantities [13]. |
| Cell Suspension Kits | Optimized kits containing enzymes and buffers for the gentle dissociation of specific tissues (e.g., tumors, brain) into high-viability single-cell suspensions, a critical first step for scRNA-seq [13]. |
| Mission Bio Tapestri | A platform designed for targeted single-cell DNA and multi-omics sequencing. The cited SDR-seq method was adapted from this technology to simultaneously profile genomic DNA loci and RNA in thousands of single cells [37]. |
Bulk and single-cell RNA-seq are not mutually exclusive technologies but are complementary tools in the modern biologist's arsenal. Bulk RNA-seq remains a powerful, cost-effective method for answering questions about population-level, averaged gene expression. Single-cell RNA-seq is indispensable for dissecting cellular heterogeneity, discovering rare cell types, and understanding complex biological systems at their fundamental resolution. The future of transcriptomics, especially in precision medicine and drug discovery, lies in strategically combining these approaches and leveraging computational integration to gain a comprehensive picture of health and disease that is both broad and deep.
Strategic Applications: Choosing the Right Tool for Your Research Goal
In the evolving landscape of genomics, bulk RNA sequencing (bulk RNA-seq) remains a foundational technology for studying gene expression across a population of cells. This method provides a population-level perspective by measuring the average gene expression profile from a tissue sample or cell population, making it an indispensable tool for numerous research and clinical applications [2] [9]. Despite the emergence of higher-resolution single-cell technologies, bulk RNA-seq continues to offer distinct advantages where a holistic view of transcriptional activity is required. Its utility is marked by cost-effectiveness, established analytical simplicity, and proven robustness in identifying global expression patterns across experimental conditions [13] [9]. This technical guide delineates the core use cases where bulk RNA-seq provides irreplaceable population-level insights, positioning it within the broader context of transcriptomics research.
Core Applications and Use Cases of Bulk RNA-seq
Bulk RNA-seq is the preferred methodology when the research question targets the collective behavior of a cell population rather than its constituent cellular diversity. Its applications are widespread across basic research, clinical diagnostics, and drug development.
Differential Gene Expression Analysis
This is one of the most prevalent applications of bulk RNA-seq. It involves comparing gene expression profiles between different experimental conditions—such as diseased versus healthy tissues, treated versus control samples, or across various time points in a time-course experiment [13] [38]. By providing an average readout, bulk RNA-seq is highly effective at identifying subtle, consistent changes in gene expression that might be obscured by technical noise in single-cell data [26]. This capability is fundamental for:
- Discovering RNA-based biomarkers and molecular signatures for diagnosis, prognosis, or patient stratification [13].
- Investigating collective changes in gene pathways and networks under various biological conditions [13].
Transcriptome Profiling of Tissues and Large Cohorts
Bulk RNA-seq is ideally suited for projects that require a global expression profile from whole tissues, organs, or large sets of samples [13]. Its cost-effectiveness and analytical straightforwardness make it feasible for:
- Large cohort studies or biobank projects where the primary goal is to link transcriptomic profiles to clinical or phenotypic data [13].
- Establishing baseline transcriptomic profiles for new or understudied organisms or tissues [13].
- Supporting deconvolution studies, where its data can be used alongside single-cell RNA-seq reference maps to infer cell type proportions in complex tissues [13].
Detection and Characterization of Novel Transcripts
Owing to its typically higher sequencing depth per sample and more comprehensive coverage of transcripts, bulk RNA-seq is a powerful tool for transcriptome annotation and discovery [2] [13]. Key applications include:
- Identifying and characterizing isoforms, alternative splicing events, gene fusions, long non-coding RNAs, and point mutations [2] [13].
- Detecting various forms of genomic alterations, including gene fusions, substitutions, and indels, sometimes revealing alterations not detected by DNA-based approaches [2]. FoundationOne Heme is a prominent example of a clinical test that leverages this multi-faceted capability for therapy selection in oncology [2].
Disease Research and Biomarker Discovery
In disease research, particularly oncology, bulk RNA-seq has broad utilities. It has been instrumental in cancer classification, the discovery of diagnostic and prognostic biomarkers, and optimizing therapeutic strategies [2] [9] [38]. For instance, comprehensive analyses of thousands of cancer samples from The Cancer Genome Atlas (TCGA) have utilized bulk RNA-seq to detect novel and clinically relevant gene fusions driving cancer pathogenesis [9].
Table 1: Key Use Cases and Supporting Methodologies for Bulk RNA-seq
| Use Case | Experimental Objective | Typical Analytical Methods |
|---|---|---|
| Differential Expression | Identify genes with significant expression changes between conditions (e.g., disease vs. healthy). | DESeq2, EdgeR [38] [39] |
| Biomarker Discovery | Find gene expression signatures correlated with diagnosis, prognosis, or treatment response. | Differential expression analysis, clustering, machine learning [2] [38] |
| Transcriptome Characterization | Annotate isoforms, detect gene fusions, and discover novel transcripts. | Cufflinks, StringTie, fusion detection algorithms (e.g., DEEPEST) [2] [40] |
| Large Cohort Profiling | Establish global expression profiles across large sample sets (e.g., population studies). | Principal Component Analysis (PCA), clustering, WGCNA [13] [4] [39] |
The Bulk RNA-seq Workflow: From Sample to Insight
A standardized workflow is crucial for generating reliable and interpretable bulk RNA-seq data. The process extends from careful experimental design through computational analysis.
Experimental Design and Sample Preparation
The process begins with careful planning. Researchers must define objectives clearly and select appropriate sample groups, ensuring the inclusion of proper controls and biological replicates to account for natural variation [38] [39]. Key steps include:
- RNA Extraction: RNA is isolated from the tissue or cell population using methods like column-based kits or TRIzol reagents. Preventing RNA degradation and contamination is critical at this stage [38].
- Quality Control: The quality of the extracted RNA is assessed using tools like a Bioanalyzer or Nanodrop. A high RNA Integrity Number (RIN) is a key indicator of sample suitability for sequencing [38] [39].
Library Preparation and Sequencing
This phase converts RNA into a format compatible with sequencing platforms.
- Reverse Transcription: RNA is converted into more stable complementary DNA (cDNA) [38].
- Fragmentation and Adapter Ligation: cDNA is fragmented into smaller pieces, and short DNA sequences (adapters) are ligated to the ends. These adapters facilitate binding to the sequencing platform and sample identification [2] [38].
- Amplification and Sequencing: The library is amplified via PCR and loaded onto a high-throughput sequencer like an Illumina system, which generates millions of short reads [38].
Bioinformatics Analysis
The raw sequencing data (FASTQ files) undergoes a multi-step computational process:
- Quality Control: Tools like FastQC evaluate read quality, followed by trimming of low-quality bases and adapters using software like Trimmomatic [40] [38].
- Read Alignment: Cleaned reads are mapped to a reference genome or transcriptome using spliced aligners such as STAR or HISAT2 [40] [38].
- Expression Quantification: Tools like featureCounts or HTSeq count the reads aligned to each gene, generating a raw count expression matrix [40] [38].
- Normalization: Raw counts are normalized using methods like TPM or DESeq2's median-of-ratios to account for differences in sequencing depth and library composition [38] [39].
- Differential Expression & Analysis: Statistical models in tools like DESeq2 or EdgeR identify significantly differentially expressed genes between conditions [38] [39]. Subsequent functional enrichment analysis (using tools like DAVID or GSEA) interprets the biological meaning of the results [38].
The following diagram illustrates the core workflow and key decision points in a bulk RNA-seq experiment.
Bulk RNA-seq vs. Single-Cell RNA-seq: A Strategic Comparison
The choice between bulk and single-cell RNA-seq is not a matter of one being superior to the other, but rather which technology is best suited to address the specific biological question at hand.
Key Differentiating Factors
- Resolution and Heterogeneity: Bulk RNA-seq provides an average expression profile, masking cellular heterogeneity. In contrast, scRNA-seq reveals distinct cell types, rare cell populations, and continuous cell states within a sample [2] [13] [9].
- Cost and Throughput: Bulk RNA-seq is significantly more affordable, typically costing about one-tenth of scRNA-seq per sample, making it practical for large-scale studies [9]. The per-sample cost of bulk is lower, and its data analysis is less computationally intensive [13] [9].
- Gene Detection Sensitivity: While scRNA-seq can struggle to detect low-abundance transcripts due to "dropout" events, bulk RNA-seq, with its pooling of RNA from many cells, often has higher sensitivity for detecting subtle expression changes of such genes [9] [26].
Choosing the Right Tool for the Question
The decision flowchart in Section 3 provides high-level guidance. The table below offers a direct comparison to aid in strategic selection.
Table 2: Strategic Comparison of Bulk RNA-seq and Single-Cell RNA-seq
| Feature | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Population average [13] [9] | Individual cell [13] [9] |
| Ideal for | Homogeneous samples, differential expression, large cohorts [13] [9] | Heterogeneous tissues, rare cell discovery, developmental trajectories [13] [9] |
| Cost per Sample | Low (~1/10th of scRNA-seq) [9] | High [9] |
| Data Complexity | Lower, more straightforward analysis [9] | High, requires specialized computational methods [9] |
| Cell Heterogeneity | Masks heterogeneity [13] [9] | Reveals heterogeneity [13] [9] |
| Rare Cell Detection | Limited, signals are diluted [9] | Possible, can identify rare populations [9] |
Successful execution of a bulk RNA-seq experiment relies on a suite of trusted reagents, kits, and software tools.
Table 3: Essential Research Reagents and Solutions for Bulk RNA-seq
| Item Category | Specific Examples | Function in Workflow |
|---|---|---|
| RNA Isolation Kits | Column-based kits (e.g., PicoPure), TRIzol reagent [38] [39] | Extract high-quality, intact total RNA from tissue or cell samples. |
| RNA QC Instruments | Bioanalyzer (Agilent), TapeStation, Nanodrop [38] [39] | Assess RNA concentration, purity, and integrity (RIN) prior to library prep. |
| Library Prep Kits | NEBNext Ultra DNA Library Prep Kit, NEBNext Poly(A) mRNA Magnetic Isolation Kit [39] | Convert RNA to sequencing-ready cDNA libraries; enrich for mRNA or deplete rRNA. |
| Sequencing Platforms | Illumina NextSeq, NovaSeq [38] | Perform high-throughput sequencing to generate raw read data (FASTQ files). |
| Alignment Software | STAR, HISAT2, TopHat2 [40] [38] | Map sequenced reads to a reference genome or transcriptome. |
| Quantification Tools | featureCounts, HTSeq [40] [38] | Count the number of reads mapped to each gene to create an expression matrix. |
| DE Analysis Packages | DESeq2, EdgeR [38] [39] | Perform statistical analysis to identify differentially expressed genes. |
Bulk RNA-seq remains a powerful, cost-effective, and robust technology for generating population-level transcriptomic insights. Its strengths in differential expression analysis, large-scale profiling, and comprehensive transcript detection ensure its continued relevance in the genomics toolbox. As the field progresses, bulk RNA-seq is not being replaced but is rather being complemented by single-cell and spatial technologies. The most powerful studies often involve an integrated approach, using bulk RNA-seq to identify large-scale expression patterns and scRNA-seq to dissect the cellular sources of those patterns [4] [24]. By understanding its key use cases and methodological principles, researchers can strategically leverage bulk RNA-seq to advance our understanding of biology and disease.
The fundamental unit of biology is the cell, and understanding the behavior of individual cells within a complex tissue is crucial for unraveling the mechanisms of development, disease, and homeostasis. For years, bulk RNA sequencing (bulk RNA-seq) served as the primary tool for transcriptome analysis, providing a population-averaged view of gene expression [13] [2]. While invaluable for identifying overall expression trends, this approach inherently masks cellular heterogeneity, as it measures the average expression profile across thousands to millions of disparate cells [41]. The advent of single-cell RNA sequencing (scRNA-seq) has revolutionized this paradigm by enabling researchers to measure gene expression in individual cells, thereby revealing the cellular diversity, identifying rare cell types, and uncovering novel cell states that drive biological processes [42] [43]. This technical guide will delineate the specific scenarios where scRNA-seq is not merely an option but a necessity, particularly when investigating heterogeneous samples. Framed within the broader comparison of bulk versus single-cell methodologies, this whitepaper provides researchers, scientists, and drug development professionals with a rigorous framework for selecting the appropriate tool to address their core biological questions.
Bulk vs. Single-Cell RNA-seq: A Technical Comparison
To make an informed choice between these two technologies, one must first understand their fundamental differences in output and capability. Bulk RNA-seq processes RNA from an entire tissue sample or cell population, resulting in a single, composite gene expression profile representing the average of all constituent cells [13] [34]. In contrast, scRNA-seq isolates individual cells, creates barcoded libraries for each, and sequences them in parallel, generating thousands of distinct transcriptome profiles from a single sample [2] [43].
Table 1: Core Technical Differences Between Bulk and Single-Cell RNA-seq
| Feature | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Population-average | Single-cell |
| Ability to Detect Heterogeneity | No, masks differences | Yes, resolves cellular diversity |
| Identification of Rare Cell Types | Limited, can be obscured | Excellent (down to <1% abundance) |
| Primary Applications | Differential expression between conditions, transcriptome annotation, splicing analysis [13] [34] | Cell type identification, developmental trajectories, tumor microenvironment mapping, rare cell discovery [13] [2] [34] |
| Typical Cost per Sample | Lower | Higher |
| Data Complexity | Lower, established analysis pipelines | High, requires specialized bioinformatics expertise [43] |
| Sample Input | Large quantity of RNA | Single cells or nuclei, requiring viable suspension [13] |
| Spatial Context | Lost (unless paired with specific techniques) | Lost during dissociation (addressed by spatial transcriptomics) [42] |
The "averaging" inherent to bulk RNA-seq becomes a significant liability in heterogeneous tissues. For instance, if a rare but disease-driving cell population constitutes only 5% of a sample, its distinctive gene expression signature will be diluted beyond detection in a bulk readout [13]. scRNA-seq circumvents this limitation, transforming the same scenario into an opportunity for discovery by enabling the isolation and characterization of that critical rare population [2].
Figure 1: A comparative workflow of Bulk versus Single-Cell RNA-seq. Bulk processing yields a single averaged profile, while single-cell processing preserves and reveals cellular heterogeneity through individual transcriptomes.
Key Applications Where Single-Cell RNA-seq Is Indispensable
The decision to employ scRNA-seq should be driven by specific biological questions centered on cellular heterogeneity. The following applications represent areas where its use is critical.
Deconvoluting Cellular Heterogeneity and Discovering Rare Cell Types
The premier application of scRNA-seq is the systematic cataloging of cell types and states within a complex tissue. This is achieved through computational clustering of cells based on their similar gene expression patterns, which can reveal previously unknown subtypes [2] [43]. A powerful example comes from cancer research, where scRNA-seq of tumor microenvironments has uncovered rare subpopulations of therapy-resistant cells that were entirely masked in bulk analyses [2]. Similarly, in immunology, scRNA-seq has refined our understanding of immune cell activation states, revealing continuous spectra of differentiation rather than discrete categories [24].
Reconstructing Developmental Lineages and Trajectories
ScRNA-seq can capture snapshots of cells in various stages of a dynamic process, such as embryonic development or stem cell differentiation. Computational methods like pseudotime analysis then order these cells along a inferred trajectory, reconstructing the sequence of transcriptional changes that occur as a cell transitions from one state to another [43] [24]. This allows researchers to identify key regulator genes and decision points in cell fate without the need for destructive time-series experiments.
Characterizing the Tumor Microenvironment (TME)
Solid tumors are complex ecosystems comprising cancer cells, immune cells, stromal cells, and vasculature. Bulk RNA-seq of a tumor homogenate cannot reliably separate the contributions of these distinct compartments. scRNA-seq, however, can dissect the TME with precision, identifying how cancer cell subclones interact with specific immune cell populations (e.g., T-cells, macrophages) to evade destruction or promote metastasis [2] [34]. This detailed cellular cartography is invaluable for developing targeted immunotherapies.
Investigating Somatic Mosaicism and Neurological Diversity
The brain is composed of an enormous diversity of cell types, and neuronal function can be highly specific to individual cells. scRNA-seq is uniquely positioned to decode this complexity, enabling the classification of neuronal and glial subtypes and linking transcriptional profiles to function [42] [41]. It is also the preferred method for studying somatic mosaicism, where genetic differences between an organism's cells drive disease.
When to Choose Bulk RNA-seq Over Single-Cell
Despite the power of scRNA-seq, bulk RNA-seq remains a vital and often more appropriate tool for many research goals. Bulk RNA-seq is the superior choice when the research question targets population-level effects rather than cell-to-cell variation [13]. Key applications include:
- Differential Gene Expression (DGE) Analysis: When comparing gene expression between distinct conditions (e.g., diseased vs. healthy tissue, treated vs. untreated), bulk RNA-seq provides a robust, cost-effective, and statistically powerful measurement of average expression changes [13] [34].
- Analyzing Large Cohorts: For biobank studies or clinical trials involving hundreds of samples, the lower cost and simpler data analysis of bulk RNA-seq make it logistically feasible [13].
- Transcriptome Annotation and Isoform Analysis: Bulk RNA-seq, especially with long-read sequencing, is highly effective for discovering novel transcripts, alternative splicing events, and gene fusions, as it provides deep, full-length coverage of the transcriptome [2].
Integrated Analysis: A Powerful Combined Approach
An emerging and powerful strategy is the integration of bulk and single-cell data [24]. In this approach, scRNA-seq data from a subset of samples is used as a high-resolution reference to "deconvolute" bulk RNA-seq data from a much larger cohort. This allows researchers to infer the proportional abundance of different cell types in the bulk samples, linking cellular composition to clinical outcomes on a scale that would be prohibitively expensive with scRNA-seq alone. A study on rheumatoid arthritis exemplified this, where scRNA-seq identified a macrophage subpopulation expressing STAT1, and bulk data analysis confirmed this gene's upregulation across a large patient cohort [24].
Experimental Protocol and Key Reagents
Executing a successful scRNA-seq experiment requires careful planning at each step. The following is a generalized protocol for a widely used droplet-based method (e.g., 10x Genomics).
Table 2: The Scientist's Toolkit: Essential Reagents and Materials for scRNA-seq
| Item | Function | Technical Considerations |
|---|---|---|
| Viable Single-Cell Suspension | The starting biological material. | High viability (>80%) and absence of clumps are critical. Achieved via enzymatic/mechanical dissociation [13] [43]. |
| Partitioning Instrument (e.g., Chromium X) | To isolate individual cells into nanoliter-scale reactions. | Creates Gel Beads-in-emulsion (GEMs) where reverse transcription occurs [13] [2]. |
| Gel Beads | Contain barcoded oligonucleotides. | Each bead has millions of oligos with a cell barcode (to tag all RNAs from one cell) and a Unique Molecular Identifier (UMI) to quantify individual transcripts [2] [43]. |
| Reverse Transcription (RT) Reagents | Convert captured mRNA into cDNA. | Occurs inside each GEM. The template-switching mechanism ensures full-length cDNA capture [43]. |
| Library Preparation Kits | Amplify and prepare barcoded cDNA for sequencing. | Must be compatible with the partitioning platform. Adds sequencing adapters [43]. |
| Bioinformatics Pipelines (e.g., Cell Ranger, Seurat) | Demultiplexing, alignment, quantification, and quality control. | Essential for processing raw data into a gene expression matrix [43] [24]. |
Detailed Step-by-Step Methodology
- Sample Preparation & Single-Cell Isolation: Tissues are dissociated into a viable single-cell suspension using optimized enzymatic or mechanical protocols. The quality of this suspension is paramount; cell viability and count must be accurately assessed [13] [43]. For frozen archives or hard-to-dissociate tissues, single-nucleus RNA-seq (snRNA-seq) is a viable alternative [43].
- Cell Partitioning and Barcoding: The suspension is loaded onto a microfluidic chip, which co-encapsulates single cells with barcoded gel beads into oil-emulsion droplets (GEMs). Within each GEM, the bead dissolves, and the cell is lysed. The released mRNA is barcoded with cell-specific barcodes and UMIs during reverse transcription [13] [2].
- cDNA Amplification and Library Construction: The barcoded cDNA from all GEMs is pooled, amplified via PCR, and fragmented to construct a sequencing library. The final library contains fragments tagged with the cell barcode, UMI, and the gene sequence [2] [43].
- Sequencing and Data Analysis: Libraries are sequenced on a high-throughput platform (e.g., Illumina). The subsequent computational analysis involves demultiplexing (assigning reads to cells), aligning sequences to a genome, and counting the unique barcoded molecules to generate a digital expression matrix for downstream biological interpretation [24].
Figure 2: A core scRNA-seq workflow diagram for a droplet-based method, outlining key steps from tissue to data.
The choice between bulk and single-cell RNA-seq is not a matter of one technology being universally superior to the other. Instead, it is a strategic decision dictated by the biological question at hand. Bulk RNA-seq is the appropriate, powerful, and efficient tool for studying population-averaged gene expression across defined conditions or large cohorts. Single-cell RNA-seq becomes indispensable when the objective is to unravel cellular heterogeneity, discover rare cell types, map developmental trajectories, or deconstruct complex tissues into their constituent parts. As the field advances, the integration of both approaches, along with emerging technologies like spatial transcriptomics and machine learning [44] [42] [45], promises a more complete and profound understanding of biology at its most fundamental level—the single cell.
In the field of transcriptomics, researchers have historically relied on two distinct approaches: bulk RNA sequencing (bulk RNA-seq) and single-cell RNA sequencing (scRNA-seq). Bulk RNA-seq provides a population-averaged gene expression profile from an entire tissue sample, making it cost-effective for identifying overall expression changes between conditions but masking cellular heterogeneity [13] [33]. In contrast, scRNA-seq reveals the transcriptional landscape of individual cells, enabling the identification of rare cell types, transient states, and cellular heterogeneity that would otherwise be averaged out in bulk measurements [13] [42]. While scRNA-seq has revolutionized our understanding of cellular diversity, it faces challenges including lower sensitivity for detecting low-abundance transcripts and higher costs associated with sequencing depth and computational analysis [13] [29].
The integration of these complementary approaches represents a paradigm shift in transcriptomic analysis. By leveraging the high-resolution cell type identification of scRNA-seq with the sensitive transcript detection of bulk RNA-seq, researchers can achieve a more complete and accurate understanding of complex biological systems [46]. This whitepaper explores the methodological framework, applications, and practical implementation of integrated bulk and single-cell RNA-seq analysis, providing researchers with a comprehensive guide to harnessing the synergistic potential of these technologies.
Methodological Framework for Data Integration
Computational Integration Strategies
The integration of bulk and single-cell transcriptomic data requires sophisticated computational approaches that leverage the specific strengths of each dataset. One powerful method is bMIND (bulk Mixture deconvolution using an Integrated Nuanced Dependence model), which utilizes scRNA-seq data as a reference to deconvolute bulk RNA-seq samples and estimate cell type-specific gene expression [46]. This approach begins by excluding genes that show minimal variation across all cell types to reduce noise. Normalized aggregate tissue-level scRNA-seq profiles for each cell type serve as the foundation for deconvolution, allowing researchers to infer cell-type proportions and expression patterns within bulk samples [46].
Another integration strategy involves using bulk RNA-seq to enhance the detection sensitivity of scRNA-seq datasets, particularly for lowly expressed and non-coding RNAs. In this approach, bulk RNA-seq data generated from fluorescence-activated cell sorting (FACS)-isolated specific cell types provides a sensitive transcript detection baseline that can be computationally integrated with scRNA-seq profiles [46]. This integration preserves the cellular specificity of scRNA-seq while significantly improving detection sensitivity for transcripts that would otherwise be missed due to the sparsity of single-cell data. The resulting hybrid dataset demonstrates enhanced accuracy in transcript detection and differential expression analysis, effectively bridging the gap between the specificity of scRNA-seq and the sensitivity of bulk RNA-seq [46].
Experimental Design for Complementary Data Generation
Effective integration often begins with thoughtful experimental design that ensures compatibility between bulk and single-cell datasets. A proven strategy involves generating matched datasets from the same biological source material. For instance, researchers can perform scRNA-seq on dissociated tissue samples while simultaneously conducting bulk RNA-seq on either flow-sorted specific cell populations or whole tissue extracts from adjacent sections [46] [24]. This parallel approach was successfully implemented in a C. elegans study that generated both scRNA-seq profiles for every anatomical neuron class and complementary bulk RNA-seq samples for 52 specific neuron types, enabling direct comparison and integration [46].
Sample preparation consistency is critical for meaningful integration. For bulk RNA-seq targeting sensitive detection of non-polyadenylated transcripts, libraries prepared with random primers rather than oligo-dT enrichment capture a broader spectrum of RNA species, including non-coding RNAs that are typically under-represented in standard scRNA-seq protocols [46]. Quality control measures should include careful assessment of potential contamination using curated lists of genes exclusively expressed outside the cell types of interest, ensuring that expression signals truly originate from the target cells [46].
Table 1: Key Computational Tools for Bulk and Single-Cell Data Integration
| Tool/Method | Primary Function | Applications | Advantages |
|---|---|---|---|
| bMIND [46] | Deconvolution of bulk RNA-seq using scRNA-seq reference | Cell type-specific expression estimation from bulk data | Accounts for nuanced dependencies between genes |
| Seurat [24] [47] | scRNA-seq analysis and integration with bulk data | Cell clustering, trajectory inference, comparative analysis | Comprehensive toolkit with extensive documentation |
| CeNGEN Integration Method [46] | Combining FACS-sorted bulk and scRNA-seq data | Enhanced sensitivity for low-abundance transcripts | Preserves cellular specificity while improving detection |
| Harmony [24] | Batch effect correction | Integrating datasets from different platforms or conditions | Preserves biological variance while removing technical artifacts |
Experimental Protocols for Integrated Studies
Protocol 1: Integrated Analysis of Neuronal Transcriptomes
This protocol, adapted from a C. elegans study [46], demonstrates how to leverage both techniques to refine transcriptomic profiles of individual neuron classes.
Sample Preparation and Sequencing:
- Generate single-cell suspensions from whole tissue samples through enzymatic or mechanical dissociation, followed by cell counting and quality control to ensure high viability and minimal clumping [13] [46].
- For scRNA-seq: Partition single cells using a microfluidic system (e.g., 10x Genomics Chromium X series) where individual cells are isolated into gel beads-in-emulsion (GEMs). Within GEMs, dissolve gel beads to release barcoded oligos, lyse cells, and capture RNA with cell-specific barcodes [13].
- For bulk RNA-seq: Isolate specific cell types using Fluorescence-Activated Cell Sorting (FACS) with cell-type-specific promoters driving fluorophore expression. Sort directly into TRIzol LS reagent [46].
- RNA Extraction: Perform chloroform extraction using Phase Lock Gel-Heavy tubes, followed by purification with Zymo-Spin IC columns. Assess RNA quality using Agilent Bioanalyzer Picochip [46].
- Library Preparation: For bulk samples, use the SoLo Ovation Ultra-Low Input RNaseq kit with modifications to optimize rRNA depletion. For scRNA-seq, follow the standard 10x Genomics library preparation protocol [46].
- Sequencing: Sequence libraries on Illumina platforms (e.g., HiSeq 2500 or NovaSeq6000) with 150 bp paired-end reads [46].
Computational Integration:
- Data Preprocessing: Map reads to the appropriate reference genome using STAR aligner. Remove duplicate reads using UMI-tools [46].
- Quality Control: Remove samples with low read counts or poor quality metrics. Mask transgene sequences (e.g., 5kb regions around promoters used in FACS labeling) to avoid artifacts [46].
- Normalization: Perform intra-sample normalization (gene length normalization for bulk samples) followed by inter-sample normalization (TMM correction in edgeR) [46].
- Data Integration: Apply the bMIND algorithm using normalized aggregate scRNA-seq profiles as a reference to deconvolute bulk RNA-seq data and enhance detection of low-abundance transcripts [46].
- Validation: Compare integrated datasets against "ground truth" gene sets with known cell-type-specific expression patterns to assess accuracy and sensitivity improvements [46].
Protocol 2: Identifying Disease-Associated Macrophage Subpopulations
This protocol, drawn from rheumatoid arthritis research [24], outlines an integrative approach to identify key cell subpopulations driving disease pathology.
Sample Collection and Processing:
- Collect synovial tissues from rheumatoid arthritis (RA) patients and healthy controls. Process immediately for single-cell analysis or snap-freeze for bulk RNA-seq [24].
- For scRNA-seq: Digest tissue pieces in enzymatic solution at 37°C for 1 hour. Filter cell suspension through 40-micron strainers and lyse red blood cells. Assess cell concentration and viability [24].
- Single-Cell Library Construction: Use the 10x Genomics Chromium Next GEM Single-Cell 3' Reagent Kit v3.1 for cell partitioning and barcoding. Reverse-transcribe RNA to cDNA and amplify to generate sequencing libraries [24].
- For bulk RNA-seq: Isolve RNA from whole tissue samples or FACS-sorted cell populations using standard protocols. Prepare libraries using poly-A selection or ribosomal RNA depletion [24].
Data Analysis and Integration:
- scRNA-seq Processing: Use Cell Ranger for alignment and quantification. Perform quality control with Seurat, filtering cells with <250 detected genes, >10% mitochondrial genes, or <500 sequencing depth. Remove doublets using DoubletFinder [24].
- Batch Correction: Integrate multiple datasets using Harmony algorithm with parameters: group.by.vars = "orig.ident", reduction.use = "pca", theta = 2, lambda = 1, sigma = 0.1 [24].
- Cell Type Identification: Cluster cells using Seurat's graph-based clustering (resolution 0.1-0.5). Annotate cell types using canonical marker genes and reference databases [24].
- Subpopulation Analysis: Extract myeloid cells and re-cluster to identify macrophage subtypes (e.g., Stat1+ macrophages, Fn1+ macrophages) using subtype-specific markers [24].
- Bulk Data Integration: Identify differentially expressed genes (DEGs) in bulk RNA-seq datasets (GSE12021, GSE55235, GSE55457, GSE77298, GSE89408) between RA and controls. Cross-reference these DEGs with marker genes from scRNA-seq subpopulations [24].
- Machine Learning Integration: Apply LASSO regression and random forest models to integrated datasets to identify key regulatory genes (e.g., STAT1) and construct prognostic models [24].
Diagram 1: Integrated Macrophage Analysis Workflow. This workflow illustrates the process of identifying disease-associated macrophage subpopulations through integrated analysis of single-cell and bulk RNA-seq data.
Applications and Validation of Integrated Approaches
Case Study 1: Refining C. elegans Neuron Transcriptomes
A landmark study demonstrating the power of integrated bulk and single-cell RNA-seq analyzed the nervous system of C. elegans, which contains 302 neurons divided into 118 anatomically distinct types [46]. Researchers generated a comprehensive scRNA-seq dataset for every neuronal class, complemented by bulk RNA-seq samples for 52 specific neuron types obtained through FACS isolation [46]. The bulk RNA-seq data, prepared with random primers to capture both poly-adenylated and non-coding RNAs, revealed transcripts that were undetectable in scRNA-seq profiles, particularly lowly expressed genes and multiple families of non-polyadenylated transcripts [46].
Computational integration of these datasets addressed fundamental limitations in both approaches. While bulk data suffered from false positives due to minimal contamination by other cell types, scRNA-seq data missed genuine low-abundance transcripts. The integrated approach preserved scRNA-seq's cellular specificity while achieving bulk-level sensitivity, resulting in a refined gene expression atlas with enhanced accuracy for differential expression analysis [46]. This integrated dataset significantly advanced the goal of delineating the molecular mechanisms defining neuronal morphology and connectivity, providing a robust framework for similar integration efforts in other organisms [46].
Case Study 2: Identifying STAT1 as a Key Regulator in Rheumatoid Arthritis
In rheumatoid arthritis research, integration of scRNA-seq and bulk RNA-seq revealed novel insights into macrophage heterogeneity and identified STAT1 as a crucial regulator of disease progression [24]. scRNA-seq analysis of 26,923 cells from synovial tissues identified distinct macrophage subpopulations, with Stat1+ macrophages significantly elevated in RA samples compared to controls [24]. Enrichment analysis demonstrated that these Stat1+ macrophages were concentrated in inflammatory pathways, suggesting their potential role in driving RA pathology [24].
Validation through integrated analysis of five bulk RNA-seq datasets (213 RA samples, 63 healthy controls) confirmed the upregulated expression of STAT1 in RA [24]. Functional experiments further established that STAT1 activation upregulated LC3 and ACSL4 while downregulating p62 and GPX4, indicating that STAT1 contributes to RA pathogenesis by modulating autophagy and ferroptosis pathways [24]. Treatment with fludarabine, a STAT1 inhibitor, reversed these changes, suggesting STAT1 and its downstream signaling pathways as potential therapeutic targets for RA [24]. This integrated approach provided not only insights into cellular heterogeneity but also a clinically relevant molecular target for therapeutic intervention.
Table 2: Key Research Reagent Solutions for Integrated Transcriptomic Studies
| Reagent/Kit | Manufacturer | Function | Application Context |
|---|---|---|---|
| Chromium Single Cell 3' Kit | 10x Genomics | Single-cell partitioning and barcoding | scRNA-seq library preparation [13] [24] |
| SoLo Ovation Ultra-Low Input RNaseq Kit | Tecan Genomics | Low-input RNA library preparation | Bulk RNA-seq from FACS-isolated cells [46] |
| TRIzol LS Reagent | Thermo Fisher | RNA preservation and stabilization | Sample collection during FACS sorting [46] |
| Zymo-Spin IC Columns | Zymo Research | RNA purification and cleanup | RNA extraction after phase separation [46] |
| Phase Lock Gel-Heavy Tubes | Quantabio | Phase separation for RNA extraction | Chloroform extraction during RNA isolation [46] |
Computational Tools and Platforms
Successful integration of bulk and single-cell RNA-seq data requires a comprehensive computational toolkit. The Seurat package (versions 4.0.0-5.0.1) serves as a cornerstone for scRNA-seq analysis, providing functions for quality control, normalization, dimensionality reduction, clustering, and differential expression analysis [24] [47]. For batch correction and integration of multiple datasets, the Harmony algorithm has proven particularly effective, successfully removing technical artifacts while preserving biological heterogeneity [24]. The monocle3 package enables pseudotime analysis and trajectory inference, allowing researchers to reconstruct developmental pathways and cellular dynamics from integrated datasets [24].
For deconvolution of bulk RNA-seq data using scRNA-seq references, the bMIND algorithm offers a sophisticated approach that accounts for nuanced dependencies between genes [46]. Alternative deconvolution methods are also available within the broader bioinformatics ecosystem. Machine learning approaches, particularly random forest models and LASSO regression, have demonstrated significant utility in identifying key genes from integrated datasets and constructing prognostic models [24] [22]. The emergence of deep learning architectures, including autoencoders and graph neural networks, further expands the analytical toolbox for extracting biological insights from complex integrated datasets [22].
Robust experimental execution requires carefully validated reagents and rigorous quality control protocols. For single-cell experiments, the 10x Genomics Chromium system provides a standardized platform for partitioning thousands of individual cells into nanoliter-scale droplets containing barcoded oligonucleotides [13] [20]. Sample preparation protocols must be optimized for specific cell types and tissues, with particular attention to dissociation techniques that preserve cell viability while minimizing stress responses [13] [32]. For challenging cell types like neutrophils, which have low mRNA levels and high RNase content, specialized preservation and processing methods are essential for generating high-quality data [32].
Quality control metrics should be established at multiple stages of the workflow. For scRNA-seq, standard QC measures include filtering cells with fewer than 250 detected genes, mitochondrial gene content exceeding 10%, or sequencing depth below 500 reads [24]. For bulk RNA-seq from sorted cells, RNA integrity should be assessed using Agilent Bioanalyzer, with careful attention to potential contamination from other cell types [46]. Ground truth validation using genes with well-established cell-type-specific expression patterns provides a critical benchmark for assessing the accuracy of integrated datasets [46].
Diagram 2: Integrated Analysis Workflow with Quality Control. This diagram outlines the key stages in integrated transcriptomic analysis, highlighting essential quality control checkpoints throughout the process.
The integration of bulk and single-cell RNA-seq data represents a transformative approach in transcriptomics, effectively bridging the gap between cellular specificity and sensitive transcript detection. As demonstrated across multiple biological contexts—from C. elegans neuroscience to rheumatoid arthritis pathophysiology—this integrated methodology provides a more complete and accurate understanding of complex biological systems than either approach could achieve independently [46] [24]. The synergistic combination of these technologies enables researchers to resolve cellular heterogeneity while maintaining sensitivity for low-abundance transcripts, ultimately accelerating discovery across basic research and translational applications.
Future advancements in integration methodologies will likely focus on several key areas. Machine learning and artificial intelligence will play an increasingly prominent role in extracting biological insights from integrated datasets, with deep learning architectures particularly suited for identifying complex patterns across multiple layers of transcriptomic data [22]. The incorporation of spatial transcriptomics technologies will add another dimension to integration efforts, preserving the spatial context that is lost in conventional scRNA-seq while maintaining single-cell resolution [42]. Additionally, multi-omics integration—combining transcriptomic data with genomic, epigenomic, and proteomic measurements—will provide increasingly comprehensive views of cellular states and functions [20] [22]. As these technologies mature and computational methods become more sophisticated and accessible, integrated approaches will undoubtedly become standard practice in transcriptomic analysis, driving innovations in both basic biological research and therapeutic development.
The transition from bulk RNA sequencing to single-cell RNA sequencing (scRNA-seq) represents a paradigm shift in drug discovery and development. While bulk RNA-seq provides a population-average view of gene expression, scRNA-seq unveils the cellular heterogeneity within tissues and tumors, revealing rare cell populations, transient states, and cell-type-specific responses that drive disease progression and therapeutic outcomes [13] [35]. This technical guide explores how scRNA-seq resolves limitations of bulk approaches and details its applications across the pharmaceutical pipeline—from target identification and validation to biomarker discovery and clinical stratification. By providing unprecedented resolution at the individual cell level, scRNA-seq enables more precise targeting of disease mechanisms, improves prediction of drug efficacy and toxicity, and facilitates development of personalized medicine approaches [35] [16].
Bulk RNA sequencing has long been a workhorse in drug discovery, providing a comprehensive snapshot of the average transcriptomic landscape across cell populations [13]. This approach enables detection of global gene expression differences between healthy and diseased samples, identification of differentially expressed pathways, and discovery of RNA-based biomarkers [18]. However, its fundamental limitation lies in averaging gene expression across all cells in a sample, potentially masking critical cell-type-specific signals and rare cellular populations that drive disease biology [13].
Single-cell RNA sequencing transcends these limitations by profiling gene expression in individual cells, revealing the cellular complexity and heterogeneity within samples [13]. This resolution is particularly valuable for understanding complex tissues like tumors, which contain diverse cell types including malignant cells, immune cells, and stromal components that interact to influence disease progression and treatment response [35] [23]. The ability to resolve this heterogeneity makes scRNA-seq uniquely powerful for identifying novel drug targets, understanding resistance mechanisms, and developing precise biomarkers [16].
Table: Comparative Analysis of Bulk RNA-seq vs. Single-Cell RNA-seq in Drug Discovery Applications
| Application | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Population average | Individual cell level |
| Heterogeneity Analysis | Masks cellular heterogeneity | Reveals rare cell populations and transient states |
| Target Identification | Identifies broadly dysregulated pathways | Pinpoints cell-type-specific drug targets |
| Biomarker Discovery | Tissue-level biomarkers | Cell-type-specific and state-specific biomarkers |
| Toxicity Assessment | Average tissue response | Identifies specific vulnerable cell types |
| Cost per Sample | Lower | Higher |
| Data Complexity | Moderate | High-dimensional, requires specialized analysis |
scRNA-seq Applications Across the Drug Discovery Pipeline
Target Identification and Validation
ScRNA-seq enables the discovery of novel drug targets by identifying genes with cell-type-specific expression patterns in disease-relevant cell populations [35]. This approach has proven particularly valuable in oncology, where it can reveal rare tumor subpopulations driving disease progression and resistance. For example, in retinoblastoma, scRNA-seq analysis of primary tumor tissues revealed distinct subpopulations of cone precursor cells, with one subpopulation (CP4) showing elevated TGF-β signaling in invasive tumors, identifying this pathway as a potential therapeutic target [23].
Beyond identifying potential targets, scRNA-seq enhances target validation through functional genomics approaches. When combined with CRISPR screening technologies, scRNA-seq enables large-scale mapping of gene function and regulatory networks at single-cell resolution [35]. Methods like Perturb-seq link genetic perturbations to transcriptomic outcomes across diverse cell types, providing comprehensive insights into gene function and potential therapeutic effects [35].
Drug Screening and Mechanism of Action Studies
Traditional drug screening approaches relying on bulk readouts like cell viability miss crucial information about cell-type-specific responses. ScRNA-seq transforms drug screening by enabling detailed profiling of how different cell populations respond to therapeutic compounds [16]. This approach is particularly powerful for understanding drug mechanisms of action, as demonstrated in a large-scale perturbation study that measured responses to 90 cytokine perturbations across 18 immune cell types, revealing both shared patterns and unique cellular behaviors that would have been undetectable in bulk analyses [16].
The technology also provides insights into drug resistance mechanisms by identifying transcriptional programs in resistant subpopulations. For instance, in B-cell acute lymphoblastic leukemia (B-ALL), combined bulk and single-cell RNA-seq analysis identified developmental states driving resistance and sensitivity to the chemotherapeutic agent asparaginase, revealing potential strategies to overcome treatment resistance [13].
Biomarker Discovery and Patient Stratification
ScRNA-seq enables the discovery of more precise biomarkers by resolving cell-type-specific expression patterns that bulk approaches average out [16]. This capability is transforming patient stratification in clinical development by identifying molecular signatures that predict treatment response [35]. For example, in colorectal cancer, scRNA-seq has led to new classifications with subtypes distinguished by unique signaling pathways, mutation profiles, and transcriptional programs, enabling more precise patient stratification and treatment selection [16].
The technology also shows promise for monitoring treatment response and disease progression. A 2024 retrospective analysis of known drug target genes demonstrated that cell-type-specific expression in disease-relevant tissues robustly predicts a target's progression from Phase I to Phase II clinical trials, highlighting scRNA-seq's potential to de-risk drug development [16].
Toxicology and Safety Assessment
ScRNA-seq enhances safety assessment by identifying specific cell types vulnerable to drug-induced toxicity, which bulk approaches might miss due to averaging effects across cell populations [48]. By profiling transcriptomic responses across all cell types in relevant tissues, scRNA-seq can reveal cell-type-specific toxicities and help understand mechanisms underlying adverse effects, enabling earlier identification of potential safety issues in the drug development process [35].
Single-Cell RNA-seq Analysis Workflow in Drug Discovery
Experimental Design and Methodological Considerations
Sample Preparation and Single-Cell Isolation
The scRNA-seq workflow begins with generating viable single-cell suspensions from biological samples through enzymatic or mechanical dissociation [13]. This critical step requires careful optimization to preserve cell viability while achieving complete dissociation, as poor-quality suspensions can significantly impact data quality. Following dissociation, cells are typically counted and assessed for viability and absence of clumps before proceeding to library preparation [13].
Single-cell isolation is achieved through various technologies, including droplet-based systems (e.g., 10x Genomics Chromium), microwell plates, or automated microfluidic devices [13] [49]. Droplet-based methods partition individual cells into nanoliter-scale droplets containing barcoded beads, where cell lysis and barcoding occur [13]. Each RNA molecule is labeled with a cell-specific barcode and unique molecular identifier (UMI) to enable accurate counting and distinguish biological signals from technical artifacts [49].
Library Preparation and Sequencing
Library preparation involves reverse transcription of barcoded RNA, cDNA amplification, and addition of sequencing adapters [49]. The specific protocol varies by platform, with methods differing in cellular throughput, molecular capture efficiency, and compatibility with various sample types [35]. For large-scale drug discovery applications, high-throughput methods like the 10x Genomics Chromium system or Parse Biosciences' combinatorial barcoding approach enable profiling of millions of cells across thousands of samples [16].
Sequencing depth requirements depend on experimental goals, with deeper sequencing needed to detect rare cell types or low-abundance transcripts [13]. For drug screening applications where detecting subtle transcriptional responses is critical, sufficient sequencing depth is essential to power differential expression analyses across cell types and conditions.
Computational Analysis Pipeline
ScRNA-seq data analysis involves multiple computational steps, beginning with pre-processing to generate count matrices where reads are assigned to cells and genes based on their barcodes [49]. Quality control filters out low-quality cells based on metrics like count depth, number of detected genes, and mitochondrial gene fraction [49]. Following normalization to address technical variations, dimensionality reduction techniques like PCA, UMAP, or t-SNE enable visualization of cellular heterogeneity [49].
Downstream analysis includes clustering cells based on transcriptional similarity, annotating cell types using marker genes, and performing differential expression testing to identify genes associated with conditions or treatments [49] [50]. For drug discovery applications, specialized analyses like trajectory inference to reconstruct cellular differentiation paths or cell-cell communication analysis to understand signaling networks provide additional biological insights [23].
Table: Key Computational Tools for scRNA-seq Analysis in Drug Discovery
| Analysis Step | Tools | Application in Drug Discovery |
|---|---|---|
| Quality Control | Cell Ranger, Scater | Filtering low-quality cells from primary tissues |
| Normalization | SCTransform, Scran | Removing technical variations across samples |
| Clustering | Seurat, Scanpy | Identifying cell populations and states |
| Differential Expression | MAST, edgeR, DESeq2 | Finding drug-responsive genes by cell type |
| Trajectory Analysis | Monocle, CytoTRACE | Modeling differentiation and treatment effects |
| Cell-Cell Communication | CellPhoneDB, NicheNet | Identifying signaling pathways for targeting |
Analytical Best Practices for Robust Drug Discovery
Differential Expression Analysis in Complex Designs
Differential gene expression (DGE) analysis in scRNA-seq requires special consideration of experimental design and statistical approaches. Unlike bulk RNA-seq, scRNA-seq data exhibits technical artifacts like dropout events (excess zeros) and high cell-to-cell variability [50]. Perhaps most importantly, cells from the same individual are not statistically independent, creating pseudoreplication that can inflate false discovery rates if not properly accounted for [50].
Current best practices recommend two main approaches for DGE analysis: pseudobulk methods that aggregate counts to the sample level followed by bulk RNA-seq tools like edgeR or DESeq2, and mixed models that incorporate random effects to account for within-sample correlations, such as MAST with random effects [50]. Both approaches outperform naive methods like Wilcoxon rank-sum test that don't account for sample-level correlations, with pseudobulk methods generally showing robust performance across diverse experimental designs [50].
Integration Across Modalities and Conditions
Drug discovery often involves comparing scRNA-seq datasets across multiple conditions, time points, or treatment groups. Batch effect correction methods like Harmony combat technical variations between experiments while preserving biological signals [23]. For more comprehensive insights, multi-omic approaches integrating scRNA-seq with other data types—such as single-cell ATAC-seq for chromatin accessibility, CRISPR perturbations for functional genomics, or spatial transcriptomics for tissue context—provide systems-level understanding of drug effects [35] [16].
The growing availability of public scRNA-seq data resources further enhances drug discovery by providing reference atlases for cell type annotation and baseline information about cellular heterogeneity in healthy and diseased tissues [35]. These resources help contextualize drug-induced changes and improve translation between model systems and human biology.
Integrative Approach for Target Identification
Case Study: Application in Retinoblastoma Research
A comprehensive analysis of scRNA-seq data from primary tumor tissues of 10 retinoblastoma patients demonstrates the power of this approach in drug discovery [23]. The study revealed distinct subpopulations of cone precursor cells with different proportions in invasive versus non-invasive tumors, highlighting cellular heterogeneity that bulk sequencing would average out [23].
Differential expression and pathway analysis identified functional diversity among cone precursor subpopulations, with the CP4 subpopulation showing elevated TGF-β signaling specifically in invasive retinoblastoma [23]. Cell-cell communication analysis further revealed rewired interaction networks in invasive tumors, with increased fibroblast–cone precursor interactions suggesting potential microenvironmental contributions to disease progression [23].
Complementary bulk RNA-seq analysis identified two molecular subtypes with distinct clinical outcomes, with subtype 1 exhibiting an immunosuppressive tumor microenvironment [23]. Through integrated analysis of both single-cell and bulk data, DOK7 was identified as a key gene associated with invasion, and functional assays confirmed its role in promoting tumor progression, validating its potential as a therapeutic target [23].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table: Key Platforms and Reagents for scRNA-seq in Drug Discovery
| Tool Category | Examples | Key Features | Drug Discovery Application |
|---|---|---|---|
| Single-Cell Platforms | 10x Genomics Chromium, Parse Biosciences Evercode | High-throughput, combinatorial barcoding | Large-scale drug screening, cohort studies |
| Library Prep Kits | 10x 3' Gene Expression, SMART-Seq2 | High sensitivity, full-length coverage | Target validation, isoform analysis |
| Analysis Software | Seurat, Scanpy, Cell Ranger | Comprehensive toolkit, scalability | Processing large drug screening datasets |
| CRISPR Screening | Perturb-seq, CROP-seq | Links genetic perturbations to transcriptomics | Target identification and validation |
| Cell Annotation | SingleR, Celldex | Automated cell type identification | Consistency across large studies |
| Spatial Transcriptomics | 10x Visium, Slide-seq | Tissue context preservation | Understanding drug distribution and effects |
Single-cell RNA sequencing has transformed from a specialized technology to an essential tool in modern drug discovery and development. By resolving cellular heterogeneity that bulk approaches inevitably average out, scRNA-seq provides unprecedented insights into disease mechanisms, therapeutic responses, and resistance pathways across the pharmaceutical pipeline. As methodologies continue to advance—increasing throughput, reducing costs, and integrating multi-omic capabilities—the application of scRNA-seq in drug discovery will expand further, enabling more targeted therapies, improved clinical success rates, and ultimately, more effective precision medicine approaches.
The identification of novel therapeutic targets in leukemia requires a deep understanding of the complex cellular heterogeneity and molecular mechanisms driving disease progression. While bulk RNA sequencing (bulk RNA-seq) has long been the standard for transcriptome analysis, it provides an averaged gene expression profile across all cells in a sample, potentially masking critical cell-type-specific signals [13]. The emergence of single-cell RNA sequencing (scRNA-seq) has revolutionized cancer research by enabling the characterization of gene expression at individual cell resolution, revealing rare cell populations and cellular states that were previously undetectable [2]. This case study examines how the integrated application of both methodologies has successfully identified and validated a druggable target in acute myeloid leukemia (AML), demonstrating their complementary value in oncology drug discovery.
Integrated Analytical Workflow
Experimental Design and Methodology
The foundational study integrated multiple sequencing approaches and analytical techniques to identify T cell senescence-related prognostic genes in AML [51]. The comprehensive workflow incorporated the following key components:
Data Acquisition and Processing:
- scRNA-seq data from dataset GSE116256 was analyzed using the Uniform Manifold Approximation and Projection (UMAP) algorithm to identify distinct cell clusters
- FindAllMarkers analysis identified differentially expressed genes (DEGs) in T-cells
- Bulk RNA-seq data from GSE114868 identified DEGs in AML versus control samples
- Clinical and survival data from The Cancer Genome Atlas AML cohort (TCGA-LAML) enabled prognostic modeling
Cross-Referencing and Validation:
- Identified DEGs were crossed with the CellAge database to pinpoint aging-related genes
- Univariate and multivariate Cox regression analyses screened for prognostic genes
- Risk models were constructed to stratify patients into high-risk and low-risk categories
- External validation utilized dataset GSE71014 to verify prognostic ability of the risk score model
- Tumor immune infiltration analysis compared tumor immune microenvironments between risk groups
- Final experimental validation employed reverse transcription quantitative polymerase chain reaction (RT-qPCR) to confirm gene expression patterns
Key Methodological Details
The scRNA-seq analysis relied on the 10x Genomics Chromium platform, which utilizes gel bead-in-emulsions (GEMs) technology to partition individual cells, with each GEM containing a cell-specific barcode to trace analytes back to their cell of origin [13]. This enabled the identification of T cell subpopulations and their specific gene expression signatures associated with senescence.
For bulk RNA-seq, standard population-level transcriptome profiling was employed, providing the average expression levels for individual genes across all cells in the sample [13]. This approach was crucial for identifying overall expression patterns that correlated with clinical outcomes across larger patient cohorts.
Table 1: Core Experimental Components in the Leukemia Target Discovery Workflow
| Component | Technology/Platform | Primary Function | Key Outcome |
|---|---|---|---|
| Single-Cell Partitioning | 10x Genomics Chromium (GEMs) | Isolate individual cells with unique barcodes | Resolution of cellular heterogeneity in T-cell populations |
| Cell Cluster Identification | UMAP Algorithm | Dimensionality reduction for cell type identification | Distinct T-cell subpopulations associated with senescence |
| Differential Expression Analysis | FindAllMarkers (Seurat) | Identify genes differentially expressed between conditions | T-cell senescence-associated genes in AML microenvironment |
| Prognostic Modeling | Cox Regression Analysis | Associate gene expression with patient survival | 4-gene prognostic signature (CALR, CDK6, HOXA9, PARP1) |
| External Validation | Independent Datasets (GSE71014) | Verify prognostic model in independent cohorts | Confirmed risk stratification accuracy across populations |
| Experimental Confirmation | RT-qPCR | Validate transcriptomic findings at bench | Verified expression patterns of prognostic genes |
Key Findings and Therapeutic Implications
Identification of Prognostic Genes and Risk Modeling
The integrated analysis identified 31 AML differentially expressed genes associated with aging, which were subsequently refined to four key prognostic genes (CALR, CDK6, HOXA9, and PARP1) through univariate, multivariate, and stepwise regression analyses [51]. These genes formed the basis of a risk model that effectively stratified AML patients into high-risk and low-risk categories with distinct survival outcomes.
The prognostic power of this 4-gene signature was validated using receiver operating characteristic (ROC) curves, which demonstrated accurate prediction of 1-, 3-, and 5-year survival in AML patients [51]. The risk model maintained its predictive accuracy when applied to external validation cohorts, supporting its robustness across diverse patient populations.
Table 2: Characteristics of Identified Prognostic Genes in AML
| Gene | Known Functions | Therapeutic Implications | Validation Method |
|---|---|---|---|
| CALR | Calreticulin protein; endoplasmic reticulum chaperone | Potential role in immunogenic cell death; possible immune modulation | RT-qPCR confirmation of expression |
| CDK6 | Cyclin-dependent kinase 6; cell cycle regulation | Established druggable target; CDK inhibitors available | Strong binding activity to target drugs predicted |
| HOXA9 | Homeobox A9 transcription factor; hematopoietic differentiation | Challenging to target directly; potential for pathway inhibition | Consistent overexpression in AML blasts |
| PARP1 | Poly(ADP-ribose) polymerase; DNA repair | FDA-approved PARP inhibitors available for other cancers | Confirmed druggability with existing therapeutic class |
Biological Significance and Druggability Assessment
The study revealed that these prognostic genes not only predicted patient outcomes but also exhibited strong binding activity to target drugs, specifically mentioning IGF1R and ABT737 [51]. This finding highlights the direct translational potential of the identified signature, suggesting that existing therapeutic agents or those in development could be repurposed for AML patients stratified by this risk model.
Tumor immune infiltration analyses further demonstrated significant differences in the tumor immune microenvironment between low- and high-risk groups, providing mechanistic insights into how these genes might influence AML progression through modulation of the immune landscape [51]. This finding aligns with the growing recognition of the importance of tumor microenvironment in leukemia pathogenesis and treatment resistance.
Signaling Pathways and Biological Mechanisms
The identified genes participate in critical oncogenic pathways that represent promising therapeutic intervention points. CALR (calreticulin) plays roles in calcium homeostasis and endoplasmic reticulum function, with emerging implications in immunogenic cell death. CDK6 is a well-established cell cycle regulator that promotes G1 to S phase transition, and its inhibition represents a validated therapeutic strategy in cancer. HOXA9 is a homeobox transcription factor involved in hematopoietic differentiation whose dysregulation is known to drive leukemogenesis. PARP1 participates in DNA repair mechanisms, and PARP inhibition induces synthetic lethality in tumors with homologous recombination deficiencies.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key Research Reagent Solutions for scRNA-seq and Bulk RNA-seq Integration
| Category | Specific Product/Platform | Application Context | Role in Workflow |
|---|---|---|---|
| Single-Cell Platform | 10x Genomics Chromium Controller | Single-cell partitioning | Creates GEMs for barcoding individual cells |
| Single-Cell Chemistry | 10x Genomics GEM-X Flex Gene Expression | High-throughput scRNA-seq | Reduces cost per cell while maintaining sensitivity |
| Cell Hashing | Anti-B2M and Anti-CD298 Antibody-Oligo Conjugates | Multiplex scRNA-seq (e.g., 96-plex) | Enables sample multiplexing and batch effect reduction |
| Bioinformatic Tools | Seurat, Cell Ranger | scRNA-seq data analysis | Cell clustering, UMAP visualization, differential expression |
| Bulk Analysis Tools | Trimmomatic, HISAT2, featureCounts | Bulk RNA-seq processing | Quality control, read alignment, gene quantification |
| Validation Reagents | RT-qPCR Assays | Gene expression validation | Confirmatory analysis of transcriptomic findings |
Discussion and Future Directions
This case study demonstrates the powerful synergy between scRNA-seq and bulk RNA-seq in advancing leukemia research toward therapeutic applications. The initial discovery of cellular heterogeneity and specific T-cell senescence patterns via scRNA-seq provided the resolution needed to identify biologically relevant processes in the AML microenvironment [51]. Subsequent validation using bulk RNA-seq across larger patient cohorts established the clinical relevance and prognostic significance of the findings, leading to a robust gene signature that could inform treatment decisions.
The integration of these technologies follows a pattern observed across oncology research, where scRNA-seq enables the decomposition of tumor ecosystems into specific cell states and rare populations, while bulk sequencing provides the statistical power for association with clinical outcomes and molecular subtypes [52] [53]. This approach is particularly valuable in leukemia, where the tumor microenvironment and immune interactions play crucial roles in disease pathogenesis and treatment response [54] [55].
Future applications of this integrated methodology will likely expand to include spatial transcriptomics, which preserves the architectural context of cells within tissues [2], and multi-omics approaches that simultaneously profile gene expression, genetic alterations, and epigenetic states at single-cell resolution. Additionally, the growing implementation of pharmacotranscriptomic profiling - combining drug screening with scRNA-seq as demonstrated in ovarian cancer [56] - holds promise for accelerating drug discovery in leukemia by directly linking transcriptional responses to therapeutic compounds.
For the drug development community, this case study highlights a viable pathway from transcriptomic discovery to target identification and validation. The four-gene signature not only stratifies patients but also points to specific druggable pathways, with CDK6 and PARP1 representing particularly promising targets given the existence of established inhibitor classes. This work exemplifies how contemporary genomics technologies can be leveraged to bridge the gap between basic cancer biology and clinical translation, ultimately contributing to more personalized and effective therapeutic strategies for leukemia patients.
Navigating Challenges: Cost, Complexity, and Data Analysis Solutions
The fundamental goal of transcriptome analysis is to understand gene expression patterns, but researchers must choose between two distinct methodological philosophies: bulk RNA sequencing (bulk RNA-seq) and single-cell RNA sequencing (scRNA-seq). These technologies represent different trade-offs between resolution, scale, cost, and technical complexity. Bulk RNA-seq provides a population-averaged view of gene expression, analyzing RNA extracted from entire tissue samples or cell populations [13] [2]. In contrast, single-cell RNA-seq resolves transcriptional heterogeneity by capturing and sequencing RNA from individual cells within a sample [41] [2]. The choice between these methods significantly impacts the biological questions that can be addressed, with bulk RNA-seq offering a "forest-level" perspective and single-cell RNA-seq revealing individual "trees" [13]. This technical guide examines the advantages, disadvantages, and appropriate applications of each method within modern biomedical research, providing researchers with a framework for selecting the optimal approach for their specific experimental needs.
Technical Foundations and Methodological Principles
Bulk RNA-Seq Workflow and Core Principles
Bulk RNA-seq functions as a population-averaging tool that measures the collective transcriptional output of all cells in a sample. The standard workflow begins with RNA extraction from homogenized tissue or cell populations, followed by conversion to cDNA and preparation of sequencing libraries [13]. This approach generates an averaged gene expression profile representing the entire cell population, making it impossible to determine whether a specific transcript originates from all cells or a specialized subset [13] [2]. The methodology assumes relative abundance comparisons, where gene expression levels are calibrated against a reference to identify up-regulated or down-regulated genes under the assumption that most genes remain unchanged across experimental conditions [57]. This foundational principle enables bulk RNA-seq to detect global expression shifts but obscures cell-to-cell variation.
Single-Cell RNA-Seq Workflow and Resolution Advantages
Single-cell RNA-seq employs fundamentally different principles to achieve cellular resolution. The workflow begins with tissue dissociation into viable single-cell suspensions, requiring careful optimization of enzymatic or mechanical dissociation protocols to maintain cell viability while preventing RNA degradation [13] [20]. The critical technological innovation involves physically separating individual cells, typically through microfluidic partitioning systems that isolate cells into nanoliter-scale reaction vessels [13] [2]. The 10x Genomics Chromium system, for instance, uses gel beads-in-emulsion (GEM) technology where each gel bead contains cell-barcoded oligos with unique molecular identifiers (UMIs) that tag individual mRNA molecules, enabling precise tracking of each transcript to its cell of origin after sequencing [13] [2]. This barcoding strategy preserves single-cell resolution throughout library preparation and sequencing, transforming our capacity to investigate cellular heterogeneity, identify rare cell populations, and characterize transitional cell states that remain inaccessible to bulk approaches [20] [41].
Workflow for Single-Cell RNA Sequencing
Comparative Analysis: Advantages and Limitations
Advantages of Bulk RNA-Seq
Bulk RNA-seq remains widely utilized due to several compelling advantages, particularly for studies requiring broad transcriptional profiling across multiple conditions or timepoints. Its most significant advantage is cost-effectiveness, both in terms of per-sample sequencing costs and reduced computational requirements for data analysis [19] [18]. The methodology benefits from established, straightforward workflows with standardized protocols and analytical pipelines that have been refined over more than a decade of use [19]. This technical maturity translates to reduced experimental complexity, as bulk RNA-seq doesn't require specialized single-cell isolation equipment or expertise in handling sensitive single-cell suspensions [13]. For projects analyzing large sample cohorts, such as clinical trials or population-level studies, bulk RNA-seq provides excellent statistical power to detect expression differences between experimental groups when biological replicates are sufficient [58]. Additionally, the technique offers superior sensitivity for detecting low-abundance transcripts within a tissue context, as the pooling of RNA from thousands of cells creates a composite signal that can reveal transcripts potentially missed in single-cell approaches due to technical limitations in capture efficiency [18].
Limitations of Bulk RNA-Seq
The primary limitation of bulk RNA-seq stems from its fundamental averaging nature, which obscures cellular heterogeneity by blending signals from potentially diverse cell types and states [13] [2]. This averaging effect can mask biologically significant patterns, particularly when rare cell populations drive critical biological processes but contribute minimally to the overall transcriptional profile [41]. In complex tissues like tumors with diverse cellular ecosystems, bulk sequencing cannot determine whether expression changes occur uniformly across all cells or are restricted to specific subpopulations [2]. This limitation extends to inability to resolve cellular dynamics, including transitional states during differentiation, immune activation, or drug response [18] [41]. The technique also suffers from sampling bias in heterogeneous tissues, where slight variations in cellular composition between samples can create apparent expression differences unrelated to the experimental condition [2]. Furthermore, bulk RNA-seq provides no capacity for novel cell type discovery, as it relies on existing knowledge about tissue composition rather than revealing previously unrecognized cellular diversity [13] [41].
Advantages of Single-Cell RNA-Seq
Single-cell RNA-seq's transformative advantage lies in its unprecedented resolution to explore transcriptional heterogeneity at the cellular level [20] [41]. This resolution enables discovery of novel cell types and states that remain invisible in bulk analyses, including rare but functionally critical populations like cancer stem cells, transitional immune cells, or unique neuronal subtypes [18] [2]. The technology permits reconstruction of developmental trajectories and cellular differentiation pathways through computational inference methods that order cells along pseudotemporal continua based on transcriptional similarity [18] [41]. This capability provides unique insights into dynamic biological processes without requiring time-series sampling. Single-cell approaches excel at dissecting complex tissues into constituent cell types and determining their proportional representation and gene expression signatures [13]. The technology also enables identification of cell-type-specific responses to perturbations, revealing how different cells within the same tissue respond to disease, treatment, or environmental changes [13] [2]. Modern scRNA-seq platforms additionally support multiomic integration, allowing simultaneous profiling of gene expression with other molecular features like chromatin accessibility, surface protein expression, or genetic variants within the same cell [18] [59].
Limitations of Single-Cell RNA-Seq
The enhanced resolution of single-cell RNA-seq comes with significant technical and practical challenges. The most commonly cited limitation is higher cost per sample, driven by specialized reagents, instrumentation, and increased sequencing depth requirements to capture rare cell types and low-expression genes [13] [41]. The methodology involves greater technical complexity throughout the workflow, particularly during sample preparation where tissue dissociation must balance cell viability with recovery of representative cell populations [13] [20]. Single-cell data presents unique analytical challenges including sparsity from excessive zero counts (both technical and biological zeros), complex normalization requirements, batch effects, and the need for specialized computational methods that differ from bulk RNA-seq approaches [57]. The technology demonstrates reduced sensitivity for lowly expressed genes in individual cells due to limited starting material and stochastic sampling effects, though this can be mitigated by sequencing more cells [57]. Current scRNA-seq methods also lose spatial context during tissue dissociation, disconnecting gene expression patterns from their original tissue architecture, though this limitation is being addressed by emerging spatial transcriptomics technologies [41]. Additionally, the field continues to grapple with statistical challenges in differential expression analysis at single-cell resolution, including proper handling of donor effects and appropriate normalization methods that preserve biological signal while removing technical artifacts [57].
Table 1: Comprehensive Comparison of Bulk vs. Single-Cell RNA-Sequencing
| Parameter | Bulk RNA-Seq | Single-Cell RNA-Seq |
|---|---|---|
| Resolution | Population-averaged [13] | Single-cell [41] |
| Cost per Sample | Lower [19] | Higher [13] |
| Technical Complexity | Moderate, established protocols [19] | High, requires specialized expertise [13] [41] |
| Data Analysis | Standardized pipelines [19] | Specialized methods for sparse data [57] |
| Sensitivity for Rare Cell Types | Limited, signals diluted [2] | Excellent, can identify rare populations [18] |
| Detection of Heterogeneity | No, averages across cells [13] | Yes, reveals cellular diversity [41] |
| Spatial Information | No, tissue homogenized [41] | No, cells dissociated (though spatial methods emerging) [41] |
| Ideal Application Scope | Differential expression between conditions, biomarker discovery, large cohort studies [13] [19] | Cell atlas construction, developmental biology, tumor heterogeneity, immune profiling [13] [2] |
| Sample Requirements | Standard RNA extraction [13] | Viable single-cell suspension [20] |
| Throughput | Typically fewer samples with more reads per sample | Thousands of cells with fewer reads per cell [2] |
Experimental Design and Method Selection Framework
Decision Framework for Method Selection
Choosing between bulk and single-cell RNA-seq requires careful consideration of research objectives, sample characteristics, and resource constraints. Bulk RNA-seq is optimal when investigating population-level responses between distinct conditions (e.g., diseased vs. healthy, treated vs. control), conducting large-scale cohort studies where cost considerations preclude single-cell analysis, performing biomarker discovery from readily accessible tissues, or studying biological systems with minimal cellular heterogeneity [13] [19]. In contrast, single-cell RNA-seq is preferred when characterizing cellular heterogeneity in complex tissues, discovering novel or rare cell populations, reconstructing developmental trajectories or lineage relationships, investigating cell-type-specific responses to perturbations, or analyzing samples where cellular composition is unknown or potentially dynamic [13] [18]. For comprehensive research programs, a sequential or integrated approach often proves most powerful, using bulk RNA-seq for initial discovery across large sample sets followed by single-cell RNA-seq to investigate specific hypotheses or timepoints at higher resolution [18] [2].
Experimental Design Decision Framework
Case Study: Integrated Approach in Cancer Research
A 2024 Cancer Cell study by Huang et al. exemplifies the powerful synergy between bulk and single-cell approaches [13]. Researchers investigated mechanisms of chemoresistance in B-cell acute lymphoblastic leukemia (B-ALL) by combining bulk RNA-seq of healthy human B cells and clinical leukemia samples with single-cell profiling. The bulk analyses identified global expression differences between sensitive and resistant samples, while single-cell RNA-seq resolved which specific developmental cell states were driving resistance versus sensitivity to asparaginase treatment [13]. This integrated methodology enabled both the detection of population-level expression patterns and the precise identification of rare resistant subpopulations that would have been averaged out in bulk-only approaches. The study demonstrates how strategically combining these technologies can accelerate therapeutic target discovery and provide insights into heterogeneous treatment responses that inform precision medicine strategies.
Essential Reagents and Research Solutions
Table 2: Key Research Reagent Solutions for RNA-Sequencing Workflows
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Cell Partitioning Systems | 10x Genomics Chromium X series [13] | Microfluidic partitioning of single cells into GEMs |
| Barcoding Beads | 10x Gel Beads [2] | Delivery of cell barcodes and UMIs to individual cells |
| Library Preparation Kits | GEM-X Flex Gene Expression assay [13] | Preparation of sequencing libraries from barcoded cDNA |
| Sample Multiplexing | GEM-X Universal 3' and 5' Multiplex assays [13] | Sample barcoding to process multiple samples in one run |
| Cell Viability Assays | Demonstrated Protocols for sample prep [13] | Assessment of cell integrity and dissociation quality |
| Enzymatic Mixes | Reverse transcription master mixes [20] | cDNA synthesis from captured mRNA |
| Analysis Software | Cell Ranger, Loupe Browser [2] | Processing, visualization, and analysis of single-cell data |
Future Perspectives and Emerging Solutions
The historical dichotomy between bulk and single-cell approaches is gradually blurring as technological advancements address current limitations. Cost reduction initiatives like the 10x Genomics GEM-X Flex assay are making single-cell profiling more accessible by lowering per-cell costs and enabling higher-throughput studies [13]. Integrated multiomic technologies now permit simultaneous profiling of gene expression with genomic variation, chromatin accessibility, or protein expression within the same single cells, providing more comprehensive molecular portraits [18] [59]. Computational method development continues to address single-cell-specific challenges like data sparsity and normalization, with emerging frameworks like GLIMES offering improved differential expression analysis by leveraging UMI counts and zero proportions within generalized mixed-effects models [57]. Spatial transcriptomics technologies are overcoming the loss of spatial context in conventional scRNA-seq by capturing gene expression information within intact tissue architecture [41]. For bulk RNA-seq, advanced analytical approaches are improving biomarker selection by focusing on genes with homogeneous expression within tumors despite inter-tumor variability, enhancing prognostic performance and clinical translation potential [2]. These converging advancements suggest that future transcriptomic research will increasingly leverage both technologies in complementary frameworks that maximize their respective strengths while mitigating their limitations.
Bulk and single-cell RNA-seq represent complementary rather than competing technologies in the transcriptomics toolkit, each with distinct advantages and optimal applications. Bulk RNA-seq remains the method of choice for hypothesis-driven research comparing defined conditions across large sample cohorts, offering cost-efficiency, established protocols, and statistical power for detecting population-level expression differences. Single-cell RNA-seq provides unparalleled resolution for discovery-phase research investigating cellular heterogeneity, rare cell populations, and dynamic biological processes, despite higher costs and technical complexity. The most impactful research strategies often integrate both approaches, using bulk sequencing to identify global expression patterns and single-cell technologies to resolve their cellular origins. As both methodologies continue to evolve, researchers are better positioned than ever to select appropriate tools—or strategic combinations thereof—to address their specific biological questions and advance our understanding of complex biological systems in health and disease.
Demystifying Single-Cell Costs and Scalability with New High-Throughput Assays
The advent of single-cell RNA sequencing (scRNA-seq) has fundamentally transformed biological research by allowing scientists to probe gene expression at the ultimate level of resolution: the individual cell. This technology has revealed critical insights into cellular heterogeneity, rare cell populations, and developmental trajectories that were previously obscured by the population-level averages generated by bulk RNA sequencing [18] [13]. While bulk RNA-seq remains a valuable, cost-effective tool for identifying global transcriptomic differences between sample groups, it inherently masks cellular diversity by providing an averaged expression profile across all cells in a sample [9]. In contrast, single-cell sequencing unlocks the ability to identify novel cell types, characterize tumor subclones, track immune cell clonal expansions, and reconstruct developmental pathways [18].
However, this revolutionary resolution has traditionally come with significant challenges in cost efficiency and technical scalability. Early single-cell methods were labor-intensive, low-throughput, and prohibitively expensive for large-scale studies. The recent development of high-throughput, microfluidics-based platforms and novel assay chemistries is fundamentally changing this landscape, making large-scale single-cell studies increasingly feasible and cost-effective. This technical guide examines the current state of single-cell sequencing costs and scalability, providing researchers with a practical framework for leveraging these advanced assays in their experimental designs.
Cost Analysis: Traditional Barriers and New Economies of Scale
Direct Cost Comparisons with Bulk Sequencing
The cost differential between bulk and single-cell RNA sequencing remains substantial, though it is narrowing with technological advancements. Bulk RNA-seq maintains a significant advantage in per-sample processing costs, typically ranging around one-tenth the cost of single-cell approaches [9]. This makes bulk sequencing the preferred method for studies requiring large sample sizes, such as cohort studies, clinical trials, or time-series experiments where population-level trends are sufficient [18].
The table below summarizes key differences between these approaches:
Table 1: Key Comparative Features of Bulk vs. Single-Cell RNA Sequencing
| Feature | Bulk RNA Sequencing | Single-Cell RNA Sequencing |
|---|---|---|
| Resolution | Population-level average [13] | Individual cell level [13] |
| Cost per Sample | Lower (~1/10 of scRNA-seq) [9] | Higher [9] |
| Cell Heterogeneity Detection | Limited [9] | High [9] |
| Rare Cell Type Detection | Limited [9] | Possible [9] |
| Gene Detection Sensitivity | Higher [9] | Lower per cell [9] |
| Data Complexity | Lower, more straightforward analysis [9] | Higher, requires specialized computational methods [9] |
| Ideal Application | Homogeneous samples, differential expression analysis [13] | Heterogeneous tissues, rare cell identification, developmental trajectories [13] |
The Evolving Cost Structure of Single-Cell Sequencing
The single-cell sequencing market is experiencing rapid growth, projected to expand from USD 3.90 billion in 2024 to USD 12.29 billion by 2032, representing a compound annual growth rate (CAGR) of 13.61% [60]. This expansion is driving competition and technological innovation that are progressively reducing costs. Several factors contribute to the changing cost structure of single-cell sequencing:
- Consumables Dominance: Consumables represent the largest cost segment, accounting for approximately 54.2% of market share in 2024 [60]. This includes essential items like assay kits, buffers, beads, and cell isolation products that are continuously consumed during research workflows.
- High-Throughput Innovations: New platforms and assay chemistries, such as 10x Genomics' GEM-X Flex Gene Expression assay, are specifically designed to reduce the cost per cell by enabling experiments with vastly higher cell numbers [13].
- Scaled Reagent Options: Manufacturers now offer smaller kit options that allow researchers to use smaller sample amounts and fewer reagents, optimizing resource utilization for pilot studies or smaller projects [13].
The strategic combination of bulk and single-cell approaches represents a powerful, cost-effective methodology. Researchers can use bulk sequencing to identify pathways of interest across many samples, then strategically employ single-cell sequencing to pinpoint which specific cells drive those changes, thus optimizing resource allocation [18].
Scalability Solutions: Experimental and Computational Advances
High-Throughput Experimental Platforms
The development of high-throughput single-cell sequencing platforms has been instrumental in addressing scalability challenges. These technologies can be broadly categorized into droplet-based and plate-based systems, each with distinct advantages for specific applications.
Table 2: High-Throughput Single-Cell Sequencing Platforms
| Technology/Platform | Core Mechanism | Throughput Capacity | Key Advantages |
|---|---|---|---|
| 10x Genomics Chromium | Droplet-based microfluidics [61] | Thousands to millions of cells [61] | High throughput, standardized, widely validated in publications [61] |
| SORT-seq | FACS sorting into 384-well plates [61] | Ideal for small populations (e.g., 380 cells/plate) [61] | Low input requirements, flexible scaling, handles large cell types [61] |
| VASA-seq | FACS sorting into 384-well plates [61] | Suitable for detailed analysis of limited cells | Full-length and total RNA sequencing, including non-coding RNAs [61] |
Recent platform innovations focus on increasing cell throughput while reducing operational complexity. The market for high-throughput single-cell sequencing platforms is growing rapidly, with an estimated value of $2.5 billion in 2025 and projected CAGR of 18% through 2033 [62]. Key developments include enhanced microfluidic designs for more efficient cell capture, improved library preparation methods for greater sensitivity, and integrated workflows that reduce manual processing steps [62].
Computational Infrastructure for Scalable Data Analysis
As single-cell experiments grow to encompass millions of cells, traditional data analysis tools face significant computational constraints. A typical single-cell experiment generates a cell-by-gene matrix with high dimensionality (20,000-50,000 genes across millions of cells), creating substantial memory and processing demands [63] [64]. Specialized computational frameworks have emerged to address these challenges:
- ScaleSC: A GPU-accelerated package that provides 20-100 times faster performance than CPU-based tools like Scanpy. It can handle datasets of 10-20 million cells on a single A100 GPU card, dramatically improving accessibility for researchers without multi-GPU systems [64].
- scSPARKL: A novel parallel analytical framework leveraging Apache Spark's distributed computing capabilities to enable efficient analysis of single-cell transcriptomic data of any size, even on commodity hardware [63].
These computational solutions implement optimized algorithms for critical processing steps including quality control, normalization, dimensionality reduction, and clustering, making terabyte-scale single-cell analysis feasible [63].
The following diagram illustrates the architecture of a distributed computing framework for single-cell data analysis:
Diagram: Distributed computing architecture for single-cell data analysis
Integrated Experimental Protocols for Scalable Single-Cell Analysis
High-Throughput Workflow for Large-Scale Studies
For studies requiring analysis of hundreds of thousands to millions of cells, an integrated experimental and computational workflow ensures robust, reproducible results:
Sample Preparation and Quality Control
- Generate high-viability single-cell suspensions using optimized dissociation protocols [13].
- Perform cell counting and viability assessment using automated systems.
- For fixed samples, consider technologies like Single Cell Gene Expression Flex that enable sequencing of formaldehyde-fixed tissues [61].
Cell Partitioning and Library Preparation
- Utilize high-throughput microfluidics systems (e.g., 10x Genomics Chromium X) for partitioning thousands of cells into nanoliter-scale reactions [13] [61].
- Implement cell barcoding strategies that enable multiplexing of multiple samples in a single run.
- Use automated liquid handling systems to reduce variability in library preparation steps.
Sequencing and Data Generation
- Optimize sequencing depth based on experimental goals (typically 20,000-50,000 reads per cell for standard gene expression profiling).
- Employ dual indexing strategies to enable sample multiplexing and reduce sequencing costs.
Computational Analysis of Large Datasets
- Implement distributed computing frameworks (e.g., scSPARKL) for processing large-scale data [63].
- Perform quality control to remove low-quality cells and doublets.
- Apply normalization and batch correction algorithms to address technical variability.
- Conduct dimensionality reduction and clustering to identify cell populations and states.
The following workflow diagram outlines the key steps in a high-throughput single-cell sequencing experiment:
Diagram: High-throughput single-cell RNA-seq workflow
Specialized Methodologies for Complex Applications
Multi-Modal Single-Cell Analysis
Advanced single-cell assays now enable simultaneous capture of multiple molecular modalities from the same cells:
- CITE-seq: Combines single-cell transcriptome analysis with surface protein quantification through antibody-derived tags [61].
- Single Cell Multiome ATAC + Gene Expression: Simultaneously profiles chromatin accessibility and gene expression in the same nuclei.
- TCR/BCR Sequencing: Characterizes immune receptor repertoire alongside transcriptomic profiles using 5' single-cell immune profiling [61].
Spatial Transcriptomics Integration
For tissues where spatial context is critical, emerging technologies enable correlation of single-cell gene expression data with histological location:
- Slide-tags Technology: A breakthrough method that spatially contextualizes single-cell sequencing data, recently commercialized by Curio Bioscience [65].
- Visium Spatial Gene Expression: Provides untargeted, genome-wide spatial mapping of gene expression while retaining tissue morphology.
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementing high-throughput single-cell sequencing requires specialized reagents and materials optimized for scalability and performance. The following table details key components of a single-cell sequencing workflow:
Table 3: Essential Research Reagent Solutions for High-Throughput Single-Cell Sequencing
| Reagent/Material | Function | Technical Considerations |
|---|---|---|
| Cell Partitioning Chips | Microfluidic chips for isolating single cells into nanoliter-scale reactions | Throughput varies by chip type (e.g., 10x Genomics Chromium chips process 500-80,000 cells) [13] |
| Master Mix Reagents | Enzyme mixtures for reverse transcription and cDNA amplification | Critical for sensitivity and detection efficiency; formulation affects library complexity [61] |
| Barcoded Gel Beads | Oligonucleotide-barcoded beads for cell-specific labeling | Each bead contains ~1 million barcodes with cell barcode, UMI, and poly(dT) sequence [13] |
| Assay Kits | Comprehensive reagent sets for specific applications (e.g., gene expression, immune profiling) | Single Cell Gene Expression Flex enables analysis of formaldehyde-fixed tissues [61] |
| Cell Viability Stains | Fluorescent dyes to distinguish live/dead cells during quality control | Critical for ensuring high-quality input material; typically used before loading cells onto chip [13] |
| Library Preparation Kits | Reagents for converting barcoded cDNA into sequencing-ready libraries | Optimized for low-input materials; include cleanup beads and amplification reagents [61] |
| Sequenceing Additives | Specialized buffers and primers to enhance sequencing performance | Improve cluster generation and read quality on Illumina platforms |
The landscape of single-cell sequencing is evolving rapidly, with high-throughput assays effectively addressing previous limitations in cost and scalability. While single-cell methods will not entirely replace bulk RNA-seq—which remains valuable for hypothesis generation and large-coort studies—the strategic integration of both approaches provides a powerful framework for comprehensive biological discovery [18]. The continuing decline in per-cell costs, coupled with computational advances that can process millions of cells, positions single-cell technologies as increasingly accessible tools for basic research, drug discovery, and clinical applications.
Looking forward, several emerging trends will further enhance scalability and application breadth: the integration of artificial intelligence for improved data interpretation, the development of more sophisticated multi-omics approaches at single-cell resolution, and continued innovation in microfluidics and sequencing chemistry. By understanding both the technical capabilities and economic considerations outlined in this guide, researchers can make informed decisions about implementing high-throughput single-cell sequencing to address their most pressing biological questions.
The choice between bulk and single-cell RNA sequencing (scRNA-seq) is fundamental to experimental design in genomics. While bulk RNA-seq provides a population-averaged transcriptome, scRNA-seq unveils cellular heterogeneity, identifying rare cell types and dynamic state transitions. However, the transition from bulk to single-cell resolution introduces significant technical hurdles, primarily in sample preparation and the preservation of cell viability. This guide details the core challenges and provides robust, current methodologies to ensure the generation of high-quality data for both approaches.
The Sample Preparation Divide: Bulk vs. Single-Cell
The initial steps of sample handling critically diverge between the two techniques, directly influencing data integrity.
Table 1: Key Differences in Sample Preparation for Bulk vs. Single-Cell RNA-seq
| Parameter | Bulk RNA-seq | Single-Cell RNA-seq (e.g., Droplet-Based) |
|---|---|---|
| Starting Input | >10,000 cells (tissue lysate or cell pellet). | 500 - 10,000 individual, viable cells. |
| Cell Lysis | Global lysis of the entire cell population. | Controlled lysis within individual partitions (droplets/wells). |
| RNA Source | Total RNA from all cells, including cytoplasmic and nuclear. | Primarily cytoplasmic (poly-A+) RNA from intact, viable cells. |
| Dominant Bias | RNA degradation, genomic DNA contamination. | Cell viability, doublet rate, batch effects, transcript capture efficiency. |
| Critical QC Metric | RNA Integrity Number (RIN > 8). | Cell Viability (>80-90%), doublet rate (<5%). |
The Centrality of Cell Viability in scRNA-seq
In bulk RNA-seq, RNA from dead cells is diluted by RNA from viable cells. In scRNA-seq, each cell's transcriptome is profiled independently. A dead cell poses two major problems:
- Technical Artifacts: Loss of cytoplasmic RNA and increased ambient RNA (from ruptured cells) that can be captured in droplets, leading to cross-contamination and a "background" signal.
- Biological Misinterpretation: Dead and dying cells exhibit aberrant transcriptomic profiles (e.g., stress responses) that can be mistaken for a genuine biological state.
Therefore, maximizing cell viability is not merely an optimization step but a prerequisite for valid scRNA-seq data.
Detailed Experimental Protocols
Protocol: Tissue Dissociation for High Viability
Objective: To isolate a single-cell suspension from solid tissue with maximal viability and minimal transcriptional stress.
Reagents:
- Hanks' Balanced Salt Solution (HBSS), cold
- Dissociation Enzyme(s) (e.g., Collagenase IV, Liberase, TrypLE)
- DNase I
- Cell Staining Buffer (PBS + 1% BSA)
- Viability Dye (e.g., Propidium Iodide, 7-AAD, DAPI) or AO/DAPI Staining Solution
Procedure:
- Collection & Mincing: Immediately after excision, place tissue in cold HBSS. Mince tissue into ~1-2 mm³ pieces using sterile scalpels on a petri dish kept on ice.
- Enzymatic Digestion: Transfer minced tissue to a tube containing pre-warmed (37°C) dissociation enzyme mix. Use a gentleMACs Dissociator or a water bath with orbital shaking for 30-45 minutes. Optimization Note: The enzyme combination and incubation time must be empirically determined for each tissue type.
- Quenching & Neutralization: Add excess cold cell staining buffer containing 10% FBS to quench enzyme activity.
- Filtration & Washing: Pass the cell suspension through a 40 μm cell strainer. Centrifuge at 300-400 x g for 5 minutes at 4°C. Resuspend the pellet in cold cell staining buffer.
- Red Blood Cell Lysis (if needed): Perform a brief ACK lysis buffer incubation (2-3 minutes) if the tissue is highly vascular. Wash immediately with excess buffer.
- DNase Treatment (if needed): If clumping is observed, resuspend pellet in buffer with 10 U/mL DNase I for 5 minutes on ice to digest free DNA.
Protocol: Cell Viability Assessment Pre-Sequencing
Objective: To accurately quantify the percentage of live cells and, if necessary, enrich for them prior to library preparation.
Method A: Fluorescence-Activated Cell Sorting (FACS)
- Staining: Resuspend ~1x10⁶ cells in 100 μL of cell staining buffer containing a viability dye (e.g., 1 μg/mL Propidium Iodide (PI) or equivalent). Incubate for 5-15 minutes on ice, protected from light.
- Analysis/Sorting: Run the sample on a flow cytometer. Live cells are identified as PI-negative. For scRNA-seq, sort the PI-negative population directly into a tube containing collection buffer (PBS + 1% BSA or 0.04% BSA). Keep samples on ice.
Method B: Automated Cell Counter with Fluorescence
- Staining: Mix 10 μL of cell suspension with 10 μL of AO/DAPI staining solution.
- Counting: Load onto a slide and analyze with an automated cell counter. Nuclei from all cells stain green with Acridine Orange (AO), while nuclei from dead/damaged cells stain blue with DAPI. The counter software calculates viability (% Viable Cells = (AO+ & DAPI- cells / Total AO+ cells) * 100).
Visualizing Workflows and Challenges
scRNA-seq Viability Workflow
Impact of Cell Viability on Data
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Sample Prep & Viability
| Item | Function | Application Context |
|---|---|---|
| Liberase TL | Enzyme blend for gentle tissue dissociation. | Superior for sensitive tissues; preserves cell surface epitopes for CITE-seq. |
| Propidium Iodide (PI) | Membrane-impermeant DNA dye. Labels dead cells. | Standard, cost-effective viability dye for flow cytometry. |
| - 7-AAD | Membrane-impermeant DNA dye. Labels dead cells. | More stable than PI, compatible with GFP channel. |
| ACD | A ready-to-use, gentle cell dissociation solution. | Ideal for dissociating adherent cell cultures into single cells. |
| MycoStrip | Rapid mycoplasma detection kit. | Essential for QC of cell cultures; mycoplasma contamination drastically alters transcriptomes. |
| Dead Cell Removal Kit | Magnetic bead-based removal of apoptotic/necrotic cells. | For enriching viability in challenging samples prior to sequencing. |
| RNAstable | Chemistry for ambient-temperature RNA stabilization. | For preserving RNA in samples during shipment or storage without freezing. |
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed molecular biology by enabling the profiling of gene expression at the resolution of individual cells, unlike bulk RNA-seq which provides an average expression profile across a population of cells [13] [26]. This unprecedented resolution allows researchers to uncover cellular heterogeneity, identify rare cell types, and characterize dynamic cellular states in complex tissues [13] [66]. However, this analytical power comes with significant technical challenges, paramount among them being the problem of data sparsity and gene dropout events [67] [68].
Gene dropouts represent a critical technical artifact in scRNA-seq data where transcripts that are actually present in a cell fail to be detected and are recorded as zero counts [67] [68]. This phenomenon stands in stark contrast to the data generated by bulk RNA-seq, where the averaging effect across thousands of cells ensures that most expressed genes are reliably detected [13]. The distinction is not merely technical but fundamentally alters the analytical landscape—while bulk RNA-seq excels at identifying global expression differences between conditions, it completely masks cell-to-cell variation [13] [18]. The dropout problem in scRNA-seq thus represents the cost of pursuing cellular-resolution insights, creating a pervasive issue that affects all subsequent biological interpretations.
This technical guide examines the molecular origins of dropout events, details the computational strategies developed to address them, and provides a framework for selecting appropriate methodologies based on specific research objectives. By contextualizing these solutions within the broader comparison of bulk versus single-cell approaches, we aim to equip researchers with the knowledge to navigate the trade-offs between resolution and data completeness in experimental design.
Understanding Dropout Events: Technical Roots and Biological Consequences
The Molecular Mechanisms Behind Dropout Events
The genesis of dropout events can be traced to the fundamental technical limitations of scRNA-seq workflows. Unlike bulk RNA-seq, which processes millions of cells simultaneously and thereby averages out many technical artifacts, scRNA-seq operates at the physical limits of detection [67]. The primary factors contributing to dropouts include:
- Low mRNA capture efficiency: The minute quantities of mRNA present in individual cells (typically ~0.1-1 pg) combined with inefficient reverse transcription reactions mean that only a fraction of transcripts are successfully converted to cDNA [68].
- Stochastic sampling effects: The limited sequencing depth per cell combined with the exponential amplification steps required for library preparation creates a scenario where low-abundance transcripts may be completely missed [67].
- Amplification biases: PCR amplification during library preparation preferentially amplifies certain transcripts, further exacerbating the under-detection of already rare molecules [68].
These technical limitations result in a characteristic zero-inflated data structure where the observed zeros in the expression matrix represent a mixture of true biological absence (biological zeros) and technical artifacts (technical zeros) [68]. Distinguishing between these two types of zeros remains a central challenge in scRNA-seq analysis.
Impact on Downstream Biological Interpretation
The consequences of dropout events permeate virtually every aspect of scRNA-seq data analysis. The Table 1 summarizes how dropouts impact key analytical applications compared to bulk RNA-seq.
Table 1: Impact of Dropout Events on Key Analytical Applications in scRNA-seq vs. Bulk RNA-seq
| Analytical Application | Impact in Bulk RNA-seq | Impact in scRNA-seq with Dropouts |
|---|---|---|
| Cell Type Identification | Not applicable (population average) | Obscured cell identities; rare populations missed [13] [66] |
| Differential Expression | Robust detection of global changes | Biased toward highly expressed genes; cell-type specific effects masked [68] |
| Trajectory Inference | Not applicable | Broken trajectories; incorrect lineage relationships [68] |
| Clustering Analysis | Not applicable | Reduced separation between clusters; inflated number of clusters [67] [68] |
| Pathway Analysis | Pathway-level insights preserved | Biased against pathways with moderate-to-lowly expressed genes [23] |
The fundamental distinction lies in how each technology handles cellular heterogeneity. Bulk RNA-seq inherently averages expression across all cells, making it robust to technical noise but blind to cellular diversity [13]. In contrast, scRNA-seq captures this diversity but at the cost of increased technical variability that can mimic or obscure true biological signals [67] [66].
Computational Frameworks for Dropout Imputation
Taxonomy of Imputation Approaches
The computational challenge of distinguishing biological zeros from technical artifacts has spawned numerous imputation methods that can be broadly categorized into four strategic frameworks:
- Model-based imputation: These methods employ statistical models, typically zero-inflated negative binomial (ZINB) distributions, to explicitly model the probability of dropout events and recover expected expression values [66].
- Data smoothing approaches: By leveraging similarities between cells or genes, these methods adjust expression values through averaging across nearest neighbors, effectively "smoothing" the expression matrix [68] [66].
- Data reconstruction techniques: Using matrix factorization or deep learning architectures, these methods reconstruct a denoised expression matrix by capturing the underlying low-dimensional structure of the data [67] [68].
- Transfer learning methods: These advanced approaches leverage external datasets, such as bulk RNA-seq or cell atlases, to guide the imputation process by incorporating prior biological knowledge [66].
The following diagram illustrates the logical relationships and workflow positioning of these four fundamental approaches within a typical scRNA-seq analysis pipeline:
Advanced Imputation Methods: From Theory to Practice
DropDAE: Denoising Autoencoder with Contrastive Learning
The DropDAE framework represents a sophisticated integration of deep learning and contrastive learning specifically designed to address dropout events [67]. The method employs a denoising autoencoder (DAE) architecture that is trained to reconstruct original expression patterns from artificially corrupted input data. The innovation of DropDAE lies in its incorporation of triplet loss—a contrastive learning technique that enhances cluster separation in the latent space [67].
The mathematical formulation of DropDAE's loss function combines reconstruction accuracy with cluster separation:
Total loss = MSE + λ × Triplet loss
Where MSE (mean squared error) ensures similarity between reconstructed and original data, and Triplet loss maximizes separation between different cell populations in the latent space [67]. This dual-objective optimization enables DropDAE to not only impute missing values but also improve downstream clustering performance.
The experimental protocol for implementing DropDAE involves:
- Input corruption: Artificially introducing additional dropout noise to the input expression matrix using parameters like
dropout.midto control dropout probability [67]. - Encoder transformation: Compressing the corrupted input through encoder layers (default: 64 units) to a bottleneck layer (default: 32 units) [67].
- Pseudo-label generation: Performing clustering on the latent representations to generate pseudo-labels for contrastive learning [67].
- Triplet selection: Sampling anchor-positive-negative triplets based on pseudo-labels for triplet loss calculation [67].
- Decoder reconstruction: Reconstructing the expression matrix through decoder layers (default: 64 units) [67].
- Parameter optimization: Updating model parameters through backpropagation with early stopping to prevent overfitting [67].
SinCWIm: Weighted Alternating Least Squares Approach
The SinCWIm method introduces a different mathematical approach based on weighted alternating least squares (WALS) that specifically addresses the varying confidence levels of different zero entries in the expression matrix [68]. Unlike methods that treat all zeros equally, SinCWIm uses Pearson correlation coefficients and hierarchical clustering to quantify the confidence of each zero entry, assigning higher weights to zeros that are more likely to be technical artifacts [68].
The SinCWIm workflow consists of four key stages:
- Data preprocessing: Filtering and normalization of the input scRNA-seq count matrix [68].
- Weight matrix calculation: Computing cell-cell similarities and assigning confidence weights to zero entries [68].
- WALS optimization: Decomposing the expression matrix using weighted alternating least squares to impute missing values [68].
- Post-processing: Applying outlier removal and data correction to ensure biological plausibility of imputed values [68].
In benchmark evaluations across eight single-cell datasets, SinCWIm demonstrated superior performance in clustering accuracy, with Adjusted Rand Index scores of 94.46% (Usoskin dataset), 96.48% (Pollen dataset), and 76.74% (Bladder dataset) [68]. The method also showed significant improvements in visualization quality and retention of differentially expressed genes [68].
Comparative Analysis of Imputation Methods
Performance Benchmarking Across Multiple Metrics
The evaluation of imputation methods requires assessment across multiple performance dimensions. Table 2 provides a comparative analysis of contemporary imputation methods based on published benchmarking studies:
Table 2: Comparative Performance of scRNA-seq Imputation Methods
| Method | Underlying Algorithm | Clustering Performance (ARI%) | Runtime Efficiency | Biological Zero Preservation | Key Strengths |
|---|---|---|---|---|---|
| SinCWIm | Weighted Alternating Least Squares | 76.74-96.48 [68] | Medium | Strong [68] | Superior clustering, differential gene retention [68] |
| DropDAE | Denoising Autoencoder + Contrastive Learning | Not reported | Lower (GPU beneficial) | Medium [67] | Enhanced cluster separation, robust representation learning [67] |
| ALRA | Truncated SVD | Variable across datasets | High | Medium [68] | Computational efficiency, stable performance [68] |
| CDSImpute | Local similarity + Multiple correlation | Lower than SinCWIm [68] | High (with small datasets) | Weak [68] | Simple implementation, fast for small datasets [68] |
| CMF-Impute | Collaborative Matrix Factorization | Lower than SinCWIm [68] | Low | Medium [68] | Integrates cell and gene similarities [68] |
The performance comparisons reveal that methods incorporating global data structure (e.g., SinCWIm, DropDAE) generally outperform local similarity-based approaches, particularly for complex datasets with high cellular heterogeneity [68]. However, this advantage comes with increased computational complexity, creating a practical trade-off between accuracy and resource requirements.
Experimental Protocols for Method Evaluation
Robust evaluation of imputation methods requires standardized experimental protocols. Based on the methodologies employed in the surveyed literature, the following protocol provides a framework for comparative assessment:
Protocol 1: Benchmarking Framework for Imputation Methods
Dataset Selection and Preprocessing
- Select diverse scRNA-seq datasets spanning different tissue types, sequencing protocols, and levels of cellular heterogeneity [68].
- Apply uniform quality control thresholds: minimum 500 genes/cell, maximum 10% mitochondrial content [66].
- Normalize using standardized approaches (e.g., log(TPM/10^5+1)) to enable fair comparisons [66].
Performance Metrics Calculation
- Clustering accuracy: Measure Adjusted Rand Index (ARI) against ground truth labels when available [68].
- Differential expression retention: Quantify the number of differentially expressed genes identified before and after imputation [68].
- Visualization quality: Assess separation and cohesion of cell populations in 2D UMAP projections [68].
- Computational efficiency: Record runtime and memory usage across different dataset sizes [67].
Validation Strategies
- Cross-validation: Employ k-fold cross-validation to assess generalizability [67].
- Biological validation: Verify that imputation results align with known biological pathways and cell-type markers [23].
- Downstream analysis impact: Evaluate how imputation affects specific applications like trajectory inference or cell-cell communication analysis [23].
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of dropout correction strategies requires both computational tools and practical experimental considerations. The following table details key resources for designing robust scRNA-seq studies:
Table 3: Essential Research Reagents and Platforms for scRNA-seq Studies
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Single-Cell Platforms | 10x Genomics Chromium X series [13] | High-throughput partitioning of single cells into GEMs for 3' or 5' gene expression library preparation [13] |
| Sample Prep Kits | GEM-X Flex Gene Expression assay [13] | Enables high-throughput single cell experiments with reduced per-cell cost and highly sensitive transcript detection [13] |
| Analysis Software | Seurat (R-based), Scanpy (Python-based) [66] | Comprehensive toolkits for QC, normalization, clustering, and differential expression analysis [66] |
| Reference Datasets | Cell atlases (e.g., Human Cell Atlas) | Provide biological priors for transfer learning imputation methods and cell type annotation [66] |
| Benchmarking Tools | scRNA-seq benchmarking pipelines | Standardized frameworks for evaluating imputation method performance across multiple metrics [68] |
Integration with Bulk RNA-seq: A Complementary Framework
Rather than viewing bulk and single-cell RNA-seq as competing technologies, the most powerful insights emerge from their strategic integration [18] [23]. Bulk RNA-seq provides a comprehensive, quantitatively accurate profile of the entire transcriptome, making it ideal for detecting subtle expression changes that might be masked by noise in single-cell data [18] [26]. Conversely, scRNA-seq reveals cellular heterogeneity and rare populations inaccessible to bulk analysis [13] [18].
This complementary relationship extends to addressing dropout events. Bulk RNA-seq data can serve as a valuable validation resource and even as a prior for transfer learning-based imputation methods [66]. The following diagram illustrates how these technologies can be integrated in a comprehensive study design:
The synergy between these approaches is particularly evident in studies like the 2024 Cancer Cell investigation by Huang et al., where both bulk and single-cell RNA-seq were leveraged to identify developmental states driving chemoresistance in B-cell acute lymphoblastic leukemia [13]. Similarly, a retinoblastoma study integrated both approaches to identify molecular subtypes and key genes associated with tumor invasion [23].
The challenge of gene dropout in single-cell RNA sequencing represents a significant but addressable barrier to extracting biological truth from high-resolution transcriptomic data. While bulk RNA-seq remains invaluable for population-level analyses and validation, the cellular heterogeneity revealed by scRNA-seq is essential for understanding complex biological systems [13] [18].
The continuing evolution of imputation methodologies—from early neighborhood-based approaches to contemporary deep learning frameworks—demonstrates the field's maturation in addressing data sparsity [67] [68]. The most promising directions include multi-omics integration, where scRNA-seq is combined with epigenetic or proteomic data to provide orthogonal validation of imputed expression values [66], and the development of conditional imputation methods that preserve true biological zeros in a cell-type-specific manner.
As single-cell technologies continue to advance, with costs decreasing and throughput increasing [69], the strategic integration of computational imputation with careful experimental design will be crucial for realizing the full potential of single-cell genomics. By embracing both the power and the limitations of scRNA-seq, and leveraging the complementary strengths of bulk approaches, researchers can navigate beyond the dropout problem toward more complete and accurate models of cellular function in health and disease.
In the context of a broader thesis contrasting bulk and single-cell RNA sequencing (scRNA-seq), understanding the advanced computational framework for bulk RNA-seq is paramount. While single-cell technologies resolve cellular heterogeneity by profiling individual cells, bulk RNA-seq provides a population-average transcriptome from a pool of cells [13]. This fundamental difference necessitates distinct computational approaches to extract biologically meaningful information from bulk data, particularly when dealing with complex tissues composed of multiple cell types. The strength of bulk RNA-seq lies in its cost-effectiveness for large cohort studies, its ability to profile samples with low cell counts or degraded RNA, and its well-established statistical frameworks for identifying expression changes between conditions [13] [70]. However, the averaging effect inherent to bulk sequencing means that shifts in the proportions of cell types can be confounded with genuine changes in gene expression within a cell type. This limitation has driven the development of sophisticated computational methods for normalization, deconvolution, and differential expression analysis, which together form the core of robust bulk RNA-seq data interpretation and bridge the conceptual gap to single-cell resolution [13] [71].
Normalization Methods: Correcting Technical Variation
Normalization is the critical first step in RNA-seq data analysis, adjusting raw count data to account for technical variations that would otherwise mask true biological signals. These technical factors include sequencing depth (the total number of reads per sample), gene length (longer genes accumulate more reads), and library composition (where a few highly expressed genes can skew the count distribution) [70] [72]. Failure to correct for these factors can lead to both false positives and false negatives in downstream analyses.
Categories and Methods of Normalization
Normalization strategies can be categorized based on their scope and purpose, as outlined in the table below.
Table 1: Categories and Methods of RNA-seq Normalization
| Category | Purpose | Key Methods | Best Use Cases |
|---|---|---|---|
| Within-Sample [72] | Compare expression of different genes within the same sample. Corrects for gene length and sequencing depth. | TPM [73] [70], FPKM/RPKM [73] [72] | Visualizing relative expression of genes within a single sample. |
| Between-Sample [72] | Compare expression of the same gene across different samples. Corrects for sequencing depth and library composition. | TMM [73] [70], RLE (used in DESeq2) [73] [70], GeTMM [73] | Differential expression analysis; required for most cross-sample comparisons. |
| Across Datasets [72] | Integrate data from multiple independent studies sequenced at different times or locations (correcting for batch effects). | Limma [72], ComBat [72] | Meta-analyses combining public datasets; multi-center studies. |
A recent benchmark study evaluating normalization methods for mapping transcriptome data onto genome-scale metabolic models (GEMs) demonstrated that between-sample methods like RLE, TMM, and GeTMM produced models with significantly lower variability and more accurately captured disease-associated genes compared to within-sample methods (TPM, FPKM) [73]. The performance of these methods can be further improved by adjusting for covariates such as patient age and gender [73].
Cellular Deconvolution: Estimating Cellular Heterogeneity
Cellular deconvolution is a powerful computational technique that addresses a core limitation of bulk RNA-seq: its inability to directly reveal the cellular composition of a tissue. By leveraging reference gene expression profiles from scRNA-seq or purified cell types, deconvolution algorithms estimate the proportional makeup of different cell types within a bulk tissue sample [71] [74]. This is crucial because observed expression changes in bulk data can be driven either by a shift in cell type abundance or by a genuine change in gene expression within a stable population.
Approaches and Benchmarking of Deconvolution Methods
Deconvolution methods are broadly classified into two categories:
- Reference-based methods require an external dataset containing cell-type-specific gene expression signatures. These methods use various statistical and machine-learning models to decompose the bulk mixture. A 2025 benchmark study using human prefrontal cortex tissue with orthogonal validation (RNAScope/ImmunoFluorescence) found Bisque and hspe (formerly dtangle) to be among the most accurate algorithms [74]. Another independent benchmarking effort also highlighted the strong performance of CIBERSORTx and MuSiC [71].
- Reference-free methods do not require a prior reference and instead infer cell-type proportions directly from the bulk data using techniques like non-negative matrix factorization (NMF). While less precise, they are valuable when high-quality reference data is unavailable [71].
The performance of these methods is highly dependent on the quality of the reference data and the choice of marker genes. The same 2025 benchmark revealed that even the best methods can show variable performance across different RNA extraction protocols and library preparation types (e.g., polyA-enrichment vs. ribosomal RNA depletion) [74].
Table 2: Key Cellular Deconvolution Algorithms
| Method | Type | Mathematical/Statistical Foundation | Input Requirements |
|---|---|---|---|
| Bisque [74] | Reference-based | Assay bias correction using linear least squares [71] | Bulk RNA-seq data + sc/snRNA-seq reference |
| hspe (dtangle) [74] | Reference-based | Linear mixing model [71] | Bulk RNA-seq data + marker genes or reference |
| CIBERSORTx [71] | Reference-based | ν-Support Vector Regression (ν-SVR) [71] | Bulk RNA-seq data + sc/snRNA-seq reference |
| MuSiC [71] | Reference-based | Weighted least squares, cross-subject scaling [71] | Bulk RNA-seq data + sc/snRNA-seq reference |
| Linseed [71] | Reference-free | Convex optimization based on simplex geometry [71] | Bulk RNA-seq data only |
| CDSeq [71] | Reference-free | Non-negative Matrix Factorization (NMF) [71] | Bulk RNA-seq data only |
Differential Expression Analysis: Identifying Significant Changes
Differential expression (DE) analysis is the cornerstone of most bulk RNA-seq studies, aiming to identify genes whose expression levels show statistically significant differences between two or more biological conditions (e.g., diseased vs. healthy, treated vs. control) [13] [6].
Experimental Design and Workflow
The reliability of DE analysis hinges on a robust experimental design. A key component is the inclusion of biological replicates—multiple independent samples per condition—which are essential for estimating biological variability and ensuring statistical power. While three replicates per condition are often considered a minimum, more may be required for studies with high inherent variability [70]. The standard workflow involves converting raw sequencing reads (FASTQ) into a gene-level count matrix, which is then used as input for statistical testing.
The following diagram illustrates the complete workflow from raw data to biological interpretation, integrating the key stages of normalization and deconvolution where applicable.
Statistical Frameworks and Tools
The count matrix generated from quantification is not directly comparable between samples due to differences in sequencing depth. After normalization using between-sample methods like TMM or RLE, statistical models are applied to test for differential expression. Two of the most widely used tools in the field are:
- DESeq2: Utilizes a negative binomial model and employs its own internal normalization method, the median-of-ratios (a type of RLE), to estimate size factors and stabilize variances before testing [70].
- limma-voom: Applies linear modeling to RNA-seq data. The "voom" method transforms the count data to log2-counts-per-million and estimates the mean-variance relationship, making the data suitable for the empirical Bayes moderation methods in the limma package [6].
These tools are considered best practice as they account for the over-dispersed nature of count data and provide robust control of false discovery rates.
The following table details key software tools and resources essential for implementing the computational workflows described in this guide.
Table 3: Essential Computational Tools for Bulk RNA-seq Analysis
| Tool / Resource | Function | Role in the Workflow |
|---|---|---|
| FastQC / MultiQC [70] | Quality Control | Assesses sequence quality, adapter contamination, and other technical metrics from FASTQ files. |
| STAR [70] [6] | Read Alignment | A splice-aware aligner that maps RNA-seq reads to a reference genome. |
| Salmon [70] [6] | Quantification | Uses pseudo-alignment for fast and accurate estimation of transcript abundances. |
| DESeq2 [73] [70] | Differential Expression | Statistical testing for DE using a negative binomial model and median-of-ratios normalization. |
| limma [6] [72] | Differential Expression / Batch Correction | A versatile package for linear modeling of RNA-seq data (with voom) and for removing batch effects. |
| Bisque [74] | Cellular Deconvolution | A reference-based algorithm for estimating cell-type proportions from bulk data. |
| hspe [74] | Cellular Deconvolution | A reference-based algorithm (formerly dtangle) known for accurate proportion estimation. |
| nf-core/rnaseq [6] | Workflow Management | A portable, community-maintained Nextflow pipeline that automates the entire data preparation workflow from FASTQ to count matrix. |
The true power of modern bulk RNA-seq analysis is realized when normalization, deconvolution, and differential expression are applied in an integrated, context-aware manner. For instance, cell type proportion estimates from deconvolution can be used as covariates in a DE analysis model to adjust for cellular heterogeneity, thereby isolating expression changes that are independent of composition shifts [71] [74]. This integrated approach allows bulk RNA-seq to answer more nuanced biological questions about complex tissues.
In conclusion, while single-cell RNA-seq offers unparalleled resolution for discovering new cell types and states, bulk RNA-seq remains a highly powerful and efficient technology for comparative transcriptomics, especially in large-scale clinical studies [13]. The advanced computational tools detailed in this guide are essential for mitigating its limitations and extracting robust, biologically interpretable results. The continued development and benchmarking of these methods, particularly in close dialogue with single-cell data, ensure that bulk RNA-seq will remain a cornerstone of functional genomics and drug development.
Synergy and Validation: Integrating Multi-Omics and Computational Advances
The journey from bulk RNA sequencing (bulk RNA-seq) to single-cell RNA sequencing (scRNA-seq) represents a fundamental shift in how researchers investigate gene expression. Traditional bulk RNA-seq provides a population-average gene expression profile, masking cellular heterogeneity and obscuring rare but critical cell populations [13] [9]. The advent of single-cell RNA sequencing has revolutionized transcriptomics by enabling researchers to probe gene expression at the individual cell level, revealing cellular diversity, novel cell types, and dynamic transitions states previously invisible to bulk methods [20] [75]. However, as the field matures, a new distinction is emerging: the division between discovery-oriented whole transcriptome approaches and hypothesis-driven targeted gene expression profiling.
This technical guide focuses on the critical transition from broad discovery to focused validation and clinical application. While whole transcriptome scRNA-seq excels at unbiased exploration, its limitations in sensitivity, cost, and scalability create barriers for translational research [29]. Targeted scRNA-seq addresses these challenges by concentrating sequencing power on predefined gene sets, transforming single-cell genomics from a discovery tool into a robust platform for clinical assay development, biomarker validation, and therapeutic monitoring [29]. By understanding this strategic transition and its technical implementation, researchers and drug development professionals can more effectively bridge the gap between biological insight and clinical impact.
Bulk RNA-seq vs. Single-Cell RNA-seq: A Foundational Comparison
To appreciate the value of targeted scRNA-seq, one must first understand the fundamental differences between bulk and single-cell approaches, and the further specialization within single-cell methodologies. The table below summarizes the key distinctions that inform methodological selection.
Table 1: Key Comparison of Bulk, Whole Transcriptome scRNA-seq, and Targeted scRNA-seq
| Feature | Bulk RNA-seq | Whole Transcriptome scRNA-seq | Targeted scRNA-seq |
|---|---|---|---|
| Resolution | Population average [13] | Individual cell [13] | Individual cell [29] |
| Cost | Lower (~1/10th of scRNA-seq) [9] | Higher [13] [9] | Cost-effective for large studies [29] |
| Data Complexity | Lower, simpler analysis [9] | Higher, requires specialized tools [9] [75] | Streamlined, focused analysis [29] |
| Cell Heterogeneity Detection | Limited, masks differences [13] [9] | High, reveals subpopulations [13] [2] | High, for predefined targets [29] |
| Rare Cell Type Detection | Limited [9] | Possible [13] [9] | Possible, if markers are targeted [29] |
| Gene Detection Sensitivity | Higher per sample [9] | Lower per cell, sparsity issues [9] [29] | Superior for targeted genes [29] |
| Primary Application | Differential expression, biomarker discovery on population level [13] | Discovery: novel cell types, heterogeneity, developmental trajectories [13] [29] | Validation, pathway interrogation, clinical assays [29] |
The progression from bulk to targeted single-cell represents a strategic refinement of goals. Bulk RNA-seq is powerful for identifying average expression differences between conditions (e.g., diseased vs. healthy tissue) but cannot determine whether these changes originate from all cells or a specific subset [13] [9]. Whole transcriptome scRNA-seq overcomes this by deconvoluting the population, enabling the discovery of new cell types and states, as demonstrated in complex tissues like tumors [2] [76]. However, its "gene dropout" problem—where low-abundance transcripts fail to be detected due to limited sequencing depth per gene—limits its quantitative accuracy for specific targets of interest [29]. Targeted scRNA-seq directly addresses this limitation by focusing resources, thereby becoming the method of choice for applications requiring sensitive and reliable quantification of a predefined gene set.
Targeted scRNA-seq: Rationale and Core Methodology
The Strategic Rationale for a Targeted Approach
Targeted scRNA-seq is defined by its focused nature: it measures the RNA expression of a pre-selected panel of genes (from dozens to hundreds) in each individual cell [29]. This approach is not intended to replace whole transcriptome methods but to complement them, creating a powerful workflow where discovery and validation inform one another.
The core advantages of targeted profiling are direct solutions to the limitations of whole transcriptome sequencing:
- Superior Sensitivity and Quantitative Accuracy: By dedicating all sequencing reads to a limited gene panel, targeted scRNA-seq achieves much deeper coverage per gene. This dramatically reduces the "gene dropout" rate, enabling reliable detection and quantification of low-abundance transcripts that are often missed in whole transcriptome assays, such as transcription factors and signaling regulators [29].
- Enhanced Cost-Effectiveness and Scalability: The reduced sequencing requirements per cell lower the cost per sample significantly. This efficiency enables the profiling of hundreds to thousands of samples, making it feasible for large-scale clinical cohort studies, high-throughput drug screens, and longitudinal monitoring [29].
- Streamlined Bioinformatics and Interpretation: Analyzing data for a few hundred genes is computationally less intensive than processing data for the entire transcriptome. This simplifies the analytical pipeline, reduces the need for advanced computational infrastructure, and makes the data more accessible for focused biological interpretation [29].
The principal trade-off is that targeted methods are blind to genes outside the panel. Therefore, their success is entirely dependent on a well-designed gene panel based on solid preliminary data from whole transcriptome studies or existing literature [29].
Experimental Workflow: From Sample to Data
The generic workflow for scRNA-seq, whether whole transcriptome or targeted, involves shared initial steps: creating a viable single-cell suspension from tissue through enzymatic or mechanical dissociation, followed by cell counting and quality control to ensure high viability and minimal debris [13] [75]. The critical divergence occurs at the library preparation stage.
Diagram: Generalized Workflow for Targeted scRNA-seq
For whole transcriptome sequencing, the barcoded cDNA is amplified and prepared for sequencing in an untargeted manner. In contrast, the targeted approach introduces a crucial step after cell lysis and barcoding: a multiplexed PCR using target-specific primers to amplify only the genes of interest. This focused amplification is the source of its enhanced sensitivity [37] [29]. Following amplification, libraries are constructed and sequenced using next-generation sequencing (NGS) platforms. The resulting data is a digital expression matrix, but unlike whole transcriptome data, it contains deep, high-fidelity information for every targeted gene in every cell.
Table 2: Essential Research Reagent Solutions for Targeted scRNA-seq
| Reagent / Material | Function | Key Considerations |
|---|---|---|
| Cell Suspension | The input material containing dissociated, viable single cells. | High viability (>80%), concentration, and absence of clumps are critical [13] [75]. |
| Fixation Reagents | Preserve cellular RNA and structure for some multi-omic assays. | Choice (e.g., PFA vs. Glyoxal) impacts gDNA and RNA quality [37]. |
| Target-Specific Primer Panels | Oligonucleotides designed to bind and amplify predefined gene targets. | Panel design is crucial; determines the scope and accuracy of the assay [29]. |
| Barcoding Beads | Microbeads containing cell-specific barcodes to tag all RNAs from a single cell. | Essential for multiplexing; allows pooling of cells [13] [2]. |
| Multiplex PCR Master Mix | Enzymes and reagents for simultaneously amplifying hundreds of gene targets. | Efficiency and fidelity directly impact sensitivity and quantitative accuracy [37]. |
| Library Preparation Kit | Prepares the amplified, barcoded products for NGS sequencing. | Optimized for the specific platform (e.g., Illumina) [75]. |
Applications in Drug Development and Clinical Translation
Targeted scRNA-seq is transforming the pharmaceutical pipeline by providing robust, scalable, and quantitative assays from early validation to clinical diagnostics. Its utility spans several critical phases of drug development.
Target Validation and Biomarker Development
While whole transcriptome scRNA-seq excels at discovering novel candidate targets and biomarkers in complex tissues, these findings require confirmation in larger, statistically powered patient cohorts [29]. Targeted scRNA-seq is the ideal tool for this validation. A candidate gene signature identified in a discovery cohort can be converted into a targeted panel to economically screen hundreds of patient samples. This confirms the biomarker's relevance and association with clinical outcomes before committing to costly development programs [2] [29]. For example, after a whole transcriptome study identifies a rare subpopulation of drug-resistant cells, a targeted panel can be designed to specifically quantify the abundance and transcriptomic state of this population across a large biobank of patient samples.
Elucidating Mechanism of Action (MoA) and Off-Target Effects
Understanding a drug's precise MoA and identifying potential off-target effects are crucial for efficacy and safety. Targeted scRNA-seq enables highly sensitive quantification of a drug's impact on its intended pathway. Researchers can treat cells with a drug candidate and use a custom panel focused on the relevant biological pathway to obtain a precise readout of on-target engagement [29]. Furthermore, by incorporating genes from known toxicity pathways (e.g., apoptosis, stress response) into the same panel, researchers can simultaneously screen for potential adverse effects, generating crucial early safety data [29].
Patient Stratification and Companion Diagnostic Development
The future of precision medicine lies in matching the right therapy to the right patient. Targeted scRNA-seq panels can be developed into clinical assays to identify patients whose tumors harbor specific cellular signatures predictive of treatment response [29]. For instance, a panel designed to quantify T-cell exhaustion markers, myeloid cell subsets, and tumor-specific antigens within the tumor microenvironment can help identify patients most likely to benefit from immunotherapy. These assays can be rigorously validated, reproducible, and cost-effective enough to be used in clinical trials for patient enrollment and, ultimately, as companion diagnostics [29].
Implementing Targeted scRNA-seq: A Practical Guide
Panel Design Strategy
The foundation of a successful targeted scRNA-seq experiment is a carefully designed gene panel. The process should begin with a clear biological or clinical question. The gene list can be derived from multiple sources: differential expression analysis of public or in-house whole transcriptome datasets, genes from a specific pathway of interest (e.g., a signaling pathway or a curated gene set), or known markers for cell types or states relevant to the disease [29]. It is also critical to include internal control genes (e.g., housekeeping genes) for data normalization. The size of the panel can range from tens to hundreds of genes, balancing the desire for comprehensive coverage with the need for maximum sequencing depth and cost-effectiveness.
An Integrated Multi-Omic Approach for Functional Phenotyping
The true power of single-cell analysis is amplified when RNA expression is linked to other molecular layers. A cutting-edge application of targeted scRNA-seq is its integration with DNA mutation profiling in the same cell. A landmark 2025 study by (Authors) demonstrated SDR-seq (single-cell DNA–RNA sequencing), a droplet-based method to simultaneously profile up to 480 genomic DNA loci and targeted RNA transcripts in thousands of single cells [37].
This multi-omic approach allows researchers to directly link a cell's genotype (e.g., a specific cancer mutation) to its functional phenotype (the resulting gene expression changes) within a complex population. For example, in a sample of B-cell lymphoma, SDR-seq was used to show that cells with a higher mutational burden exhibited elevated expression of genes involved in B-cell receptor signaling and tumorigenesis [37]. This ability to unambiguously connect cause and effect at single-cell resolution provides a powerful platform for dissecting disease mechanisms and therapy resistance.
Diagram: Multi-Omic Targeted Profiling Connects Genotype to Phenotype
Data Analysis and Quality Control
While less computationally intensive than whole transcriptome analysis, targeted scRNA-seq data still requires rigorous quality control. Standard scRNA-seq QC metrics apply, including filtering cells based on the number of detected genes and the percentage of reads mapping to mitochondrial genes [77] [78]. For targeted data, additional specific metrics should be evaluated:
- Panel Specificity: The vast majority of reads should map to the targeted genes.
- Amplification Uniformity: Check that all targeted genes are efficiently amplified without significant bias.
- Sensitivity: Assess the detection rate for expected low-abundance targets.
Following QC, analysis typically involves cell clustering based on the expression of the targeted gene panel and subsequent differential expression analysis to compare conditions (e.g., pre- vs. post-treatment) within defined cell subpopulations [76].
The evolution of transcriptomics from bulk to single-cell resolution has unveiled a new dimension of biological complexity. The subsequent specialization into whole transcriptome and targeted scRNA-seq represents a maturation of the field, acknowledging that different research questions demand tailored tools. Whole transcriptome sequencing remains the undisputed champion for exploratory discovery and atlas-building. However, for the critical path that leads from a biological discovery to a validated target, a robust clinical assay, or an effective therapeutic, targeted scRNA-seq is the indispensable tool.
Its superior sensitivity, cost-effectiveness, and scalability make it uniquely suited to meet the stringent demands of translational research and drug development. By providing a quantitative and focused readout of disease-relevant pathways across large patient cohorts, targeted scRNA-seq is poised to accelerate the development of precision medicine, ensuring that groundbreaking discoveries at the bench are effectively translated into impactful applications at the bedside.
The fundamental difference between bulk and single-cell RNA sequencing (RNA-seq) technologies creates both a challenge and an opportunity for modern genomic research. Bulk RNA-seq provides a population-level average of gene expression across all cells in a sample, functioning like a "smoothie" where individual cellular ingredients become blended beyond recognition [13]. In contrast, single-cell RNA sequencing (scRNA-seq) profiles the complete transcriptome of each individual cell, preserving the unique identity of every cellular "ingredient" and revealing the heterogeneity within tissues [13]. This resolution gap has driven the development of computational deconvolution methods—mathematical approaches that infer cell-type-specific signals from bulk data using single-cell references.
The practical importance of deconvolution extends across biomedical research and drug development. For disease research, understanding how cell type proportions change in diseased versus healthy tissues can identify key cellular drivers of pathology. In drug development, deconvolution can determine whether a therapeutic effect operates through specific cell types, enabling more targeted therapies. Additionally, deconvolution breathes new life into vast repositories of existing bulk RNA-seq data collected over past decades, allowing re-analysis through a cellular resolution lens previously unavailable [79].
Core Computational Challenges in Deconvolution
Despite conceptual appeal, deconvolution faces significant biological and technical hurdles. A primary challenge stems from transcriptome size variation across different cell types. Cells naturally contain different total numbers of RNA molecules—for instance, red blood cells predominantly express hemoglobin genes while stem cells may express thousands of genes [80]. Traditional normalization methods like Counts Per 10 Thousand (CP10K) assume equal transcriptome sizes across cell types, artificially inflating the apparent expression of cell types with smaller transcriptomes and distorting downstream deconvolution [79].
The gene length effect presents another key challenge. Bulk RNA-seq protocols typically produce reads proportional to gene length, while single-cell methods using Unique Molecular Identifiers (UMIs) are length-agnostic [79]. This fundamental technical difference creates systematic biases when using scRNA-seq data as a reference for bulk deconvolution.
Additionally, biological variability in gene expression within cell types creates discrepancies between reference and target datasets. Current methods often utilize average expression values while ignoring expression variances, discarding potentially valuable information about the distribution of gene expression within cell populations [79].
The Reference Data Dilemma
Deconvolution methods fundamentally divide into reference-based and reference-free approaches, each with distinct advantages and limitations [71]. Reference-based methods require single-cell or purified cell-type expression profiles as references to estimate cellular proportions in bulk data. These methods generally provide higher accuracy when reliable reference data exists but are vulnerable to batch effects and technical differences between reference and target datasets [71] [81]. Reference-free methods infer compositions without external references using statistical patterns within the bulk data itself, offering flexibility when reference data is unavailable but typically providing less precise cell type identification [71].
Current Methodological Landscape
Algorithmic Approaches and Classifications
The computational landscape for deconvolution has diversified substantially, with methods employing distinct mathematical foundations:
Table 1: Classification of Deconvolution Methods by Computational Approach
| Method Category | Representative Tools | Core Mathematical Foundation | Reference Requirement |
|---|---|---|---|
| Regression-Based | CIBERSORTx, MuSiC, Bisque, DWLS | Support vector regression, weighted least squares, constrained optimization | Reference-based |
| Probabilistic Models | BayesPrism, cell2location, RCTD | Bayesian inference, negative binomial models, topic modeling | Reference-based |
| Matrix Factorization | CDSeq, TOAST, GS-NMF | Non-negative matrix factorization (NMF), convex optimization | Reference-free |
| Deep Learning | DAISM-DNN, sNucConv, OmicsTweezer | Neural networks, autoencoders, optimal transport | Both types |
| Enrichment-Based | xCell, ESTIMATE | Gene set enrichment analysis, rank-based statistics | Marker genes only |
Regression-based methods like MuSiC and Bisque establish themselves as top performers in independent benchmarks. MuSiC employs weighted least squares regression leveraging cross-subject scRNA-seq data, while Bisque implements gene-specific bulk expression transformations to correct for technology-derived biases [74] [82]. CIBERSORTx extends support vector regression to deconvolution, providing both cell fraction estimates and imputed cell-type-specific gene expression profiles [71].
Probabilistic frameworks like BayesPrism apply Bayesian models to account for technical noise and biological heterogeneity, often demonstrating superior performance with complex tissues like tumors [74]. Emerging deep learning approaches like OmicsTweezer integrate optimal transport theory with neural networks to align simulated and real bulk data in a shared latent space, effectively mitigating batch effects and distribution shifts across platforms [81].
Experimental Workflow for Deconvolution Studies
The following diagram illustrates the standard experimental workflow for conducting deconvolution studies, from data collection through validation:
Performance Benchmarking Insights
Independent benchmarking studies provide crucial guidance for method selection. A comprehensive 2025 evaluation using human prefrontal cortex data with orthogonal validation found Bisque and hspe (formerly dtangle) to be the most accurate methods across multiple conditions [74]. This multi-assay dataset included bulk RNA-seq, single-nucleus RNA-seq (snRNA-seq), and RNAScope/immunofluorescence measurements from the same tissue blocks, providing rare ground-truth validation.
Table 2: Benchmarking Results of Leading Deconvolution Methods (2025)
| Method | Mathematical Foundation | Performance Ranking | Key Strengths | Implementation |
|---|---|---|---|---|
| Bisque | Regression with assay bias correction | 1st | Handles technical differences between assays | R |
| hspe (dtangle) | Linear mixing model | 1st | Robust marker selection, minimal bias | R |
| MuSiC | Weighted least squares | 2nd | Effective for closely related cell types | R |
| BayesPrism | Bayesian modeling | 3rd | Handles biological heterogeneity | R/Python |
| CIBERSORTx | Support vector regression | 4th | Cell-type-specific expression imputation | Web, R |
| DWLS | Weighted least squares | 5th | Suitable for low-resolution references | R |
Performance variations across studies highlight the importance of context-specific method selection. A robustness assessment found reference-based methods generally outperform reference-free approaches when high-quality reference data exists, while reference-free methods provide viable alternatives when suitable references are unavailable [71].
Advanced Techniques and Emerging Solutions
Addressing Fundamental Limitations
Recent methodological innovations target core deconvolution challenges. ReDeconv introduces Count based on Linearized Transcriptome Size (CLTS) normalization, specifically addressing transcriptome size variation by preserving biological differences in RNA content across cell types while removing technology-derived effects [79] [80]. This approach corrects the scaling effects that cause traditional methods to misidentify differentially expressed genes and misestimate rare cell type proportions.
The sNucConv framework addresses tissues where single-cell dissociation is problematic, such as adipose tissue with large adipocytes. By incorporating per-gene and cell-type-specific corrections trained on single-nucleus RNA-seq data, sNucConv achieves exceptional accuracy (R∼0.9) for human adipose tissue deconvolution [83].
Generative methods like sc-CMGAN (stepwise Generative Adversarial Network based on cell markers) augment scarce single-cell reference data through synthetic data generation. This approach demonstrates particular value for addressing subject heterogeneity and reference data shortages, significantly improving deconvolution accuracy across multiple datasets and methods [82].
Spatial Transcriptomics Deconvolution
The emergence of spatial transcriptomics technologies creates new deconvolution challenges and opportunities. While providing spatial context, most platforms have limited resolution, with capture spots containing multiple cells. Computational deconvolution methods for spatial data include:
- Probabilistic models (cell2location, STRIDE, SpatialDecon) using Bayesian inference to model underlying data distributions
- Non-negative matrix factorization approaches (NMFreg, SPOTlight) decomposing spatial expression matrices
- Graph-based methods (DSTG, SD2) leveraging spatial relationships between spots
- Optimal transport techniques (SpaOTsc) aligning single-cell and spatial distributions [84]
These methods enable the construction of high-resolution cellular maps from spatially barcoded data, revealing tissue organization principles and microenvironmental interactions in health and disease.
Practical Implementation Framework
Experimental Design Considerations
Successful deconvolution requires careful experimental planning. Reference data quality fundamentally limits deconvolution accuracy. Ideally, reference single-cell data should derive from the same tissue type and condition as the target bulk data, with sufficient cellular coverage of all relevant cell types. When exact matches are unavailable, cross-tissue references may be used with appropriate validation.
Bulk RNA-seq protocol selection significantly impacts deconvolution performance. PolyA-selected and ribosomal RNA-depleted protocols capture different RNA populations, with polyA enrichment showing higher exonic mapping rates and RiboZero methods capturing more diverse gene biotypes including non-polyadenylated transcripts [74]. The deconvolution reference must be compatible with the target bulk data's RNA population.
Marker gene selection represents another critical analytical choice. Methods like the novel Mean Ratio approach identify genes expressed in target cell types with minimal expression in non-target types, outperforming traditional differential expression methods for deconvolution tasks [74].
Table 3: Research Reagent Solutions for Deconvolution Studies
| Tool/Resource | Type | Function | Access |
|---|---|---|---|
| ReDeconv | Algorithmic tool | Corrects transcriptome size variation in normalization | https://redeconv.stjude.org |
| DeconvoBuddies | R/Bioconductor package | Provides benchmark datasets & marker selection methods | Bioconductor |
| Single-cell reference atlases | Data resource | Cell-type-specific expression references | CZI CellxGene, Human Cell Atlas |
| OmicsTweezer | Unified framework | Multi-omics deconvolution with batch effect correction | Python package |
| Scaden | Deep learning tool | Pretrained models for specific tissues | Python package |
Validation Strategies
Rigorous validation remains essential for credible deconvolution results. Orthogonal approaches include:
- Histological validation using immunohistochemistry or RNAscope from adjacent tissue sections
- Flow cytometry or mass cytometry on matched samples
- Pseudobulk validation by aggregating single-cell data from held-out samples
- Multi-method consensus comparing results across independent algorithms
The creation of multi-assay datasets with orthogonal proportion measurements, like the human prefrontal cortex dataset [74], provides invaluable resources for validation and method development.
Future Directions and Clinical Applications
Emerging Methodological Trends
The deconvolution field continues evolving along several trajectories. Multi-omics integration approaches like OmicsTweezer enable deconvolution across transcriptomic, proteomic, and epigenomic data types, providing complementary views of cellular composition [81]. Context-specific deconvolution frameworks like CLIER (Pathway-Level Information ExtractoR) extract single-cell-informed signatures from bulk data tailored to specific biological contexts [85].
The integration of generative artificial intelligence continues to advance, with methods like sc-CMGAN demonstrating how carefully engineered synthetic data can address fundamental limitations of real-world single-cell datasets [82]. Distribution-independent models that robustly handle batch effects and platform differences will be crucial for applying deconvolution to heterogeneous clinical datasets.
Clinical Translation in Drug Development
For drug development professionals, deconvolution offers powerful applications for target identification and patient stratification. By analyzing clinical trial bulk RNA-seq data, researchers can:
- Identify cell type proportion changes associated with treatment response
- Discover cellular targets of drug activity
- Develop biomarkers based on cellular composition shifts
- Understand tumor microenvironment remodeling during therapy
As methods continue improving, computational deconvolution will increasingly bridge the resolution gap between practical bulk profiling and the cellular heterogeneity underlying disease mechanisms and therapeutic responses.
Computational deconvolution represents an essential methodology for extracting cellular insights from bulk genomic data, effectively bridging the technological divide between scalable population-level measurements and informative single-cell resolution. As methods continue addressing fundamental challenges like transcriptome size variation, batch effects, and reference data limitations, deconvolution will play an increasingly central role in both basic research and translational applications. For researchers and drug development professionals, understanding these capabilities and limitations enables more effective deployment of deconvolution approaches to uncover biologically meaningful and clinically actionable insights from bulk RNA-seq data.
The transition from bulk RNA sequencing (RNA-seq) to single-cell RNA sequencing (scRNA-seq) represents a paradigm shift in transcriptomic research. While bulk RNA-seq measures average gene expression across cell populations, obscuring cellular heterogeneity, scRNA-seq enables the resolution of gene expression at the individual cell level, revealing rare cell types, dynamic transitions, and complex cellular ecosystems [86] [87]. This technological evolution has been driven by two principal high-throughput approaches: droplet-based microfluidics and combinatorial barcoding. Understanding their comparative performance is crucial for researchers designing studies in immunology, oncology, and developmental biology. This review provides a technical benchmarking of these platforms, framing the discussion within the broader context of moving beyond population-averaged measurements toward true single-cell resolution.
Core Technological Principles and Workflows
Droplet-Based Microfluidics Systems
Droplet-based technologies, commercialized notably by 10x Genomics, utilize microfluidic chips to partition individual cells into nanoliter-scale water-in-oil droplets, each functioning as an isolated reaction chamber [88] [87]. The core innovation lies in the Gel Bead-in-Emulsion (GEM) system. Each gel bead is coated with millions of oligonucleotides containing poly(dT) sequences for mRNA capture, a cell barcode unique to each bead, and a unique molecular identifier (UMI) for each mRNA molecule [87]. Within each droplet, cells are lysed, mRNA binds to the bead's primers, and reverse transcription occurs, tagging all cDNA from a single cell with the same barcode. After breaking the emulsion, the pooled cDNA is amplified and prepared for sequencing, with computational demultiplexing assigning reads to their cell of origin based on the barcode [87].
Combinatorial Barcoding Systems
Combinatorial barcoding technologies, such as the SPLiT-seq protocol commercialized by Parse Biosciences, take a fundamentally different approach that does not require physical partitioning of cells [86] [89]. Instead, cells are fixed and permeabilized, and the entire population is processed in suspension. The method relies on multiple successive rounds of split-pool barcoding [86]. Cells are distributed into a multi-well plate, where each well contains a unique well-specific barcode that is appended to cellular transcripts via in-cell reverse transcription. After each round, cells are pooled and then randomly split into a new plate for the next round of barcoding. After several rounds (typically 3-4), each cell receives a unique combination of barcodes that serves as its cellular identifier [86] [89]. This process labels cells in situ, using the cells themselves as reaction vessels, thereby avoiding the need for microfluidic encapsulation.
Workflow Visualization
The following diagram illustrates the key procedural differences between these two foundational approaches.
Technical Benchmarking: A Quantitative Comparison
Direct comparative studies reveal distinct performance profiles for each platform, impacting their suitability for different research scenarios. A 2024 benchmark study using human Peripheral Blood Mononuclear Cells (PBMCs) from healthy donors provides a direct, quantitative comparison [86].
Table 1: Performance Metrics for Droplet vs. Combinatorial Barcoding Platforms
| Performance Metric | 10x Genomics (Droplet-Based) | Parse Biosciences (Combinatorial Barcoding) |
|---|---|---|
| Cell Capture Efficiency | ~53% of input cells recovered [86] | ~27% of input cells recovered [86] |
| Gene Detection Sensitivity | Median ~1,900 genes/cell (at 20k reads/cell) [86] | Median ~2,300 genes/cell (at 20k reads/cell) [86] |
| Library Efficiency (Valid Barcodes) | ~98% of reads [86] | ~85% of reads [86] |
| Read Distribution (Exonic/Intronic) | Higher exonic reads (oligo-dT bias) [86] | Higher intronic reads (mixed priming) [86] |
| Multiplexing Capacity | Limited per lane; uses cell hashing [88] | High; native multiplexing for up to 96+ samples [86] |
| Doublet/Multiplet Rate | Typically <5% with optimal loading [87] | Can be minimized via combinatorial complexity [86] |
| Key Technical Advantage | High cell throughput, established workflow | Superior gene detection, flexible sample scaling |
Analysis of Key Performance Differences
- Sensitivity vs. Capture Efficiency: Parse's combinatorial barcoding demonstrates a ~1.2-fold increase in gene detection sensitivity compared to 10x Genomics, despite a lower cell capture efficiency [86]. This higher sensitivity is crucial for identifying rare cell types and subtle transcriptional states.
- Library and Priming Bias: The difference in read distribution—higher exonic reads in 10x versus higher intronic reads in Parse—stems from their reverse transcription chemistries. 10x uses oligo-dT primers, creating a 3'-bias, while Parse utilizes a mixture of oligo-dT and random hexamers, providing a more uniform coverage that can capture nascent transcripts [86].
- Scalability and Multiplexing: A defining feature of combinatorial barcoding is its innate capacity for high-level multiplexing. The first round of barcoding in a 96-well plate allows 96 samples to be processed in a single experiment, dramatically reducing batch effects and per-sample costs [86]. Droplet-based systems typically process one sample per channel, though techniques like Cell Hashing can be used to multiplex a limited number of samples [88].
Experimental Design and Benchmarking Methodologies
Robust benchmarking of scRNA-seq platforms requires carefully controlled experiments and standardized metrics.
Standard Validation Experiments
- Species-Mixing Experiments: This is the gold-standard technique for quantifying doublet rates. Human and mouse cells are mixed in known ratios and processed together. Bioinformatic tools can then easily identify heterotypic doublets (droplets containing one human and one mouse cell) based on their mixed-species expression profile [88]. The observed heterotypic doublet rate is used to estimate the total doublet rate, including homotypic doublets (two cells of the same species) [88].
- Analysis of Built-In Truths: Using well-characterized sample types, such as PBMCs from healthy donors, provides a "ground truth" for expected cell types and their known marker genes [86] [90]. The accuracy of a platform is assessed by its ability to recapitulate these known biological states without introducing technical artifacts.
- Spike-In Controls: The use of synthetic RNA controls, such as those from the External RNA Control Consortium (ERCC), allows for the assessment of technical sensitivity, dynamic range, and quantification accuracy across platforms [90].
Key Metrics for Assessment
When evaluating data quality, researchers should focus on several key metrics derived from the raw data and initial processing:
- Cell Recovery and Doublet Rate: The number of cells recovered compared to input and the fraction of multiplets, which confound biological interpretation.
- Sequencing Saturation: Indicates the comprehensiveness of transcript capture.
- Genes and UMIs per Cell: Measures the depth of transcriptomic profiling for each cell.
- Mitochondrial Read Percentage: A high percentage can indicate poor cell viability during processing.
The Scientist's Toolkit: Essential Reagents and Solutions
Table 2: Key Research Reagents and Their Functions in scRNA-seq Workflows
| Reagent / Solution | Core Function | Technology Context |
|---|---|---|
| Barcoded Gel Beads | Deliver cell barcode and UMI via oligo(dT) for mRNA capture. | Essential for droplet-based systems (10x Genomics) [87]. |
| Fixation and Permeabilization Buffers | Maintain cellular RNA content while allowing reagent entry. | Critical for combinatorial barcoding; enables multi-round barcoding on fixed cells [86] [89]. |
| Partitioning Oil & Microfluidic Chips | Generate stable, monodisperse droplets for single-cell isolation. | Core consumables for droplet-based platforms [87]. |
| Reverse Transcription Mix | Convert captured mRNA into stable, barcoded cDNA. | Common to both technologies; enzyme efficiency impacts sensitivity. |
| Cell Hashing Antibodies | Tag cells with sample-specific barcoded antibodies for multiplexing. | Used to pool samples in droplet-based systems pre-encapsulation [88]. |
| Template Switching Oligo (TSO) | Enable full-length cDNA synthesis independent of poly(A) tails. | Used in some protocols to reduce 3' bias [87]. |
| Exonuclease I | Degrade unused primers post-reverse transcription to reduce background. | Common clean-up step in library preparation. |
Application-Oriented Platform Selection
The choice between droplet-based and combinatorial barcoding technologies is not a matter of superiority but of aligning platform strengths with specific research goals and constraints.
Optimal Use Cases for Droplet-Based Platforms
- Large-Scale Cell Atlas Projects: When the goal is to profile a very large number of cells (tens to hundreds of thousands) from a limited number of samples, the high cell throughput and efficiency of 10x Genomics is advantageous [87].
- Integrated Multi-omic Profiling: Droplet-based systems offer well-established, commercialized workflows for combined gene expression and epigenetic analysis (ATAC-seq), cell surface protein quantification (CITE-seq), or immune receptor sequencing [91].
- Studies Requiring Fresh Cells: The workflow is optimized for fresh, live cell suspensions.
Optimal Use Cases for Combinatorial Barcoding Platforms
- Large-Scale, Multi-Sample Cohort Studies: The native ability to multiplex 96 or more samples in a single kit is ideal for longitudinal studies, drug screens, or large patient cohorts, as it drastically reduces batch effects and per-sample cost [86] [89].
- Studies with Challenging or Fragile Cell Types: The absence of microfluidic chambers makes Parse more suitable for large cells (e.g., activated macrophages), fragile cells (e.g., granulocytes), or cells prone to aggregation, which can clog droplet systems [89].
- Biobanked or Fixed Samples: The compatibility with fixed, permeabilized cells allows for the analysis of archived samples and provides scheduling flexibility, as the barcoding steps can be paused [86].
The benchmarking of droplet-based and combinatorial barcoding scRNA-seq platforms reveals a clear trade-off between high cell capture efficiency and high multiplexing capacity with superior gene sensitivity. The droplet-based 10x Genomics platform remains a robust, high-throughput choice for projects focused on deep characterization of a few samples. In contrast, combinatorial barcoding from Parse Biosciences offers a transformative model for studies requiring massive sample multiplexing, analysis of challenging cell types, or work with fixed specimens.
Looking ahead, the field is moving toward hybrid technologies that combine the strengths of both approaches. For instance, UDA-seq integrates an initial droplet-based barcoding step with a second round of combinatorial indexing, dramatically increasing throughput while maintaining data quality [91]. Furthermore, the integration of scRNA-seq with other modalities—such as spatial transcriptomics, proteomics, and epigenetics—will continue to be a major frontier. As these technologies evolve and costs decrease, the comprehensive, single-cell dissection of biological systems will become standard practice, accelerating discovery in basic research and therapeutic development.
The evolution of RNA sequencing (RNA-seq) technologies has fundamentally transformed biological research, moving from bulk RNA-seq that provides population-averaged gene expression profiles to single-cell RNA sequencing (scRNA-seq) that resolves transcriptional heterogeneity at the individual cell level [13] [2]. While bulk RNA-seq remains valuable for differential gene expression analysis and biomarker discovery in homogeneous populations, it masks cellular diversity and cannot resolve rare cell types or transient states [13] [9]. scRNA-seq has broken this barrier, enabling the discovery of novel cell types, characterization of developmental trajectories, and dissection of complex tissues like tumors [13] [2] [92].
The natural progression beyond transcriptomics alone lies in multi-omics integration—combining scRNA-seq with genomic, epigenomic, proteomic, and spatial data to build a comprehensive understanding of cellular function and regulation [20] [93] [92]. This integrated approach provides unprecedented insights into the molecular mechanisms driving health and disease, particularly in complex biological systems like the tumor microenvironment [2] [92]. This technical guide explores the methodologies, applications, and computational frameworks for successfully integrating single-cell transcriptomics with other data modalities, with special emphasis on spatial context and drug discovery applications.
Technical Foundations: Bulk RNA-seq vs. Single-Cell RNA-seq
Fundamental Methodological Differences
Bulk RNA-seq and scRNA-seq differ fundamentally in their experimental approaches, resolution capabilities, and analytical requirements. The table below summarizes the core distinctions between these two technologies:
Table 1: Key Technical Differences Between Bulk and Single-Cell RNA Sequencing
| Feature | Bulk RNA-Seq | Single-Cell RNA-Seq |
|---|---|---|
| Resolution | Population average [13] | Individual cell level [13] [9] |
| Cell Heterogeneity Detection | Limited; masks diversity [13] [9] | High; reveals cellular subtypes [13] [9] |
| Cost per Sample | Lower (~$300/sample) [9] | Higher (~$500-$2000/sample) [9] |
| Sample Input | Higher RNA requirements [9] | Single cells or low RNA input [9] |
| Rare Cell Detection | Limited; signals diluted [9] | Possible; identifies rare populations [9] |
| Data Complexity | Lower; simpler analysis [13] [9] | Higher; specialized computational methods [13] [9] [49] |
| Primary Applications | Differential expression, biomarker discovery [13] | Cell atlas construction, heterogeneity studies, developmental trajectories [13] [29] |
Single-Cell RNA-Seq Workflow and Quality Considerations
The scRNA-seq workflow involves critical steps that differ significantly from bulk approaches. After tissue dissociation into viable single-cell suspensions, individual cells are partitioned using microfluidic devices (e.g., 10x Genomics Chromium system) where each cell is encapsulated in a droplet with a barcoded bead [13] [2]. Following cell lysis, mRNA is barcoded with cell-specific barcodes and unique molecular identifiers (UMIs) to track individual transcripts and mitigate amplification biases [2] [94].
Quality control is paramount in scRNA-seq experiments. Key QC metrics include:
- Number of counts per barcode: Filters low-quality cells or empty droplets
- Number of genes per barcode: Identifies multiplets (high gene count) or poor-quality cells (low gene count)
- Fraction of mitochondrial counts: High percentages indicate stressed or dying cells [49] [94]
The computational analysis pipeline involves normalization to account for varying sequencing depth, feature selection to identify highly variable genes, dimensionality reduction (PCA, t-SNE, UMAP), and clustering to identify cell populations [49] [94]. Cell types are then annotated based on marker gene expression, followed by downstream analysis such as differential expression and trajectory inference.
Multi-Omics Integration Methodologies
Spatial Integration with SIMO
The Spatial Integration of Multi-Omics (SIMO) computational method represents a significant advancement for integrating scRNA-seq with spatial transcriptomics (ST) and other modalities [93]. SIMO uses a sequential mapping process that first integrates ST data with scRNA-seq data based on transcriptomic similarity, then maps non-transcriptomic single-cell data (e.g., scATAC-seq) through an optimal transport algorithm [93].
The SIMO workflow involves:
- Initial Transcriptomic Mapping: Integration of ST and scRNA-seq using k-nearest neighbor (k-NN) graphs and fused Gromov-Wasserstein optimal transport
- Cross-Modality Bridging: Using gene activity scores derived from scATAC-seq to link epigenetic and transcriptomic data
- Spatial Allocation: Precisely allocating single-cell data to specific spatial locations based on modality similarity [93]
Diagram: SIMO Computational Workflow for Multi-Omics Spatial Integration
Combined Analysis of scRNA-seq and Bulk RNA-seq
Integrating scRNA-seq with bulk RNA-seq leverages the strengths of both approaches. The single-cell data resolves cellular heterogeneity, while bulk sequencing provides a broader transcriptome overview often with higher sensitivity for low-expression genes [9] [24]. A practical application of this integration is demonstrated in rheumatoid arthritis research, where both technologies were combined to characterize macrophage heterogeneity [24].
The methodology for this integrated analysis includes:
- Cell Type Deconvolution: Using scRNA-seq data as a reference to estimate cell type proportions from bulk RNA-seq data
- Differential Expression Analysis: Identifying expression changes both between cell types (via scRNA-seq) and across conditions (via bulk RNA-seq)
- Key Driver Identification: Applying machine learning models (LASSO regression, random forests) to prioritize candidate genes from integrated datasets [24]
In the rheumatoid arthritis study, this integrated approach identified STAT1 as a key regulator in pro-inflammatory macrophages, which was validated through functional experiments showing its role in modulating autophagy and ferroptosis pathways [24].
Targeted Single-Cell Profiling for Drug Development
While whole transcriptome scRNA-seq is ideal for discovery-phase research, targeted gene expression profiling offers advantages for applied drug development. Targeted approaches focus sequencing resources on a predefined gene set, providing superior sensitivity for detecting low-abundance transcripts and enabling more cost-effective large-scale studies [29].
Table 2: Comparison of Whole Transcriptome vs. Targeted scRNA-seq Approaches
| Characteristic | Whole Transcriptome | Targeted Approach |
|---|---|---|
| Gene Coverage | All genes (~20,000) [29] | Predefined panel (dozens to thousands) [29] |
| Sensitivity | Lower; gene dropout issues [29] | Higher; focused sequencing depth [29] |
| Cost per Cell | Higher [29] | Lower [29] |
| Primary Applications | Discovery research, cell atlas construction [29] | Target validation, biomarker assays, clinical translation [29] |
| Data Complexity | High; requires specialized bioinformatics [29] | Lower; more streamlined analysis [29] |
| Therapeutic Context | Target identification [29] [92] | Mechanism of action studies, patient stratification [29] [92] |
Targeted scRNA-seq panels are particularly valuable for:
- Target Validation: Confirming candidate target expression across large patient cohorts
- Mechanism of Action Studies: Quantifying pathway engagement after drug treatment
- Biomarker Development: Creating clinically applicable assays for patient stratification [29] [92]
Applications in Drug Discovery and Development
Target Identification and Validation
scRNA-seq has revolutionized target discovery by enabling the identification of cell-type-specific disease drivers that were previously masked in bulk analyses [92]. In cancer research, scRNA-seq has revealed rare subpopulations of treatment-resistant cells and identified their specific expression signatures [2] [92]. For example, in melanoma, a minor cell population expressing high levels of AXL was found to develop resistance to RAF and MEK inhibitors [2]. Similarly, in breast cancer, scRNA-seq identified drug-tolerant-specific RNA variants that were absent in treatment-naive cells [2].
The integration of scRNA-seq with genomic data enables the direct linking of mutations to transcriptional consequences within the same cell [29] [92]. This is particularly valuable for understanding resistance mechanisms and identifying combinatorial targets to overcome resistance.
Mechanism of Action and Biomarker Analysis
Multi-omics approaches provide powerful insights into drug mechanisms by capturing simultaneous changes across molecular layers. The application of these methods in immunology and oncology has revealed how therapeutics remodel the tumor microenvironment and alter cell-cell communication [2] [92].
Diagram: Multi-Omics in Drug Discovery Pipeline
For biomarker development, scRNA-seq identifies cell-type-specific expression signatures that can be translated into targeted panels for clinical use [29] [92]. This approach was used in acute myeloid leukemia, where immune cell biosignatures predicting treatment response were converted into targeted panels for patient selection [29].
Studying Drug Resistance and Adverse Effects
Longitudinal scRNA-seq studies of tumor evolution during therapy have provided unprecedented insights into resistance mechanisms [2] [92]. By analyzing patient samples before, during, and after treatment, researchers can track the expansion of resistant subclones and identify their vulnerabilities [92].
Additionally, multi-omics approaches help elucidate adverse drug reactions by identifying cell-type-specific toxicities and pathway perturbations [92]. This is particularly valuable in immuno-oncology, where understanding the cellular players in immune-related adverse events can guide toxicity management and drug combination strategies.
Practical Implementation and Research Solutions
Experimental Design Considerations
Successful multi-omics studies require careful experimental planning. Key considerations include:
- Sample Preparation: Optimization of dissociation protocols to maintain cell viability while preserving RNA integrity [20] [49]
- Cell Number Determination: Balancing sequencing depth with the number of cells profiled to adequately capture rare populations [49] [94]
- Replication: Including sufficient biological replicates to account for donor-to-donor variability [49]
- Controls: Incorporating positive controls and spike-in RNAs to monitor technical variability [49]
For spatial multi-omics, additional considerations include:
- Spatial Resolution: Matching single-cell data to appropriate spatial resolution technologies
- Registration Accuracy: Ensuring proper alignment between different molecular modalities [93]
Computational Tools and Pipelines
The analysis of multi-omics data requires specialized computational tools. Key resources include:
- Seurat: Comprehensive R toolkit for single-cell genomics [49] [24]
- Scanpy: Python-based single-cell analysis suite [49]
- SIMO: Spatial integration of multi-omics datasets [93]
- Monocle3: Trajectory inference and analysis [24]
- Cell Ranger: 10x Genomics pipeline for processing scRNA-seq data [94]
Quality control should follow established best practices, including filtering based on UMI counts, detected genes, and mitochondrial percentage [49] [94]. Batch effect correction methods such as Harmony are essential when integrating datasets from different sources or experimental batches [24].
Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Multi-Omics Studies
| Reagent/Platform | Function | Application Context |
|---|---|---|
| 10x Genomics Chromium | Single-cell partitioning [13] [2] | High-throughput scRNA-seq and multi-omics |
| Gel Beads with Barcodes | Cell barcoding and mRNA capture [13] [2] | Unique labeling of individual cell transcripts |
| Unique Molecular Identifiers (UMIs) | Molecular counting and quantification [2] [94] | Accurate transcript quantification eliminating PCR bias |
| Feature Barcoding Oligos | Antibody-derived tag detection [94] | Cellular protein surface marker quantification |
| Chromium X Series Instruments | Microfluidic partitioning system [13] | Automated single-cell encapsulation |
| Nuclei Isolation Kits | Nuclear RNA preparation [20] | snRNA-seq for frozen or difficult-to-dissociate tissues |
| Spatial Transcriptomics Slides | Spatial RNA profiling [93] | Tissue context preservation for spatial omics |
Future Perspectives and Challenges
The multi-omics field continues to evolve rapidly, with several emerging trends shaping its future. Spatial multi-omics technologies are advancing toward subcellular resolution while maintaining tissue context [93]. Computational methods are increasingly incorporating machine learning and artificial intelligence to extract patterns from high-dimensional multi-omics datasets [20] [92]. The integration of single-cell epigenomics (ATAC-seq, DNA methylation) with transcriptomics provides insights into gene regulatory mechanisms underlying cellular heterogeneity [20] [93].
Remaining challenges include:
- Technical Artifacts: Batch effects, platform-specific biases, and sample preparation variability
- Data Integration: Developing robust mathematical frameworks for combining disparate data types
- Scalability: Managing computational demands of large-scale multi-omics studies
- Clinical Translation: Establishing standardized protocols for diagnostic applications [20] [92]
As these challenges are addressed, multi-omics approaches will become increasingly central to both basic research and translational applications, particularly in precision medicine and drug development.
The integration of scRNA-seq with genomic, spatial, and other molecular data represents the cutting edge of biological research. This multi-omics approach enables a comprehensive understanding of cellular heterogeneity, tissue organization, and disease mechanisms that was previously unattainable with bulk sequencing methods. While technical and computational challenges remain, the continued development of experimental technologies and analytical frameworks is rapidly advancing the field. For researchers and drug development professionals, mastering these multi-omics approaches is increasingly essential for driving innovation in basic science and therapeutic development.
The Role of AI and Machine Learning in Analyzing High-Dimensional Transcriptomic Data
The advent of high-throughput transcriptomic technologies has revolutionized molecular biology, generating unprecedented volumes of data that present both opportunities and analytical challenges. The field has evolved from bulk RNA sequencing (bulk RNA-seq), which provides a population-average gene expression profile, to single-cell RNA sequencing (scRNA-seq), which resolves expression at the individual cell level [13]. This progression has dramatically increased data dimensionality—where a bulk RNA-seq experiment might yield a single expression value per gene across a sample, a scRNA-seq experiment can generate expression profiles for thousands of genes across tens of thousands of individual cells [95]. This high-dimensional data contains critical biological information about cellular heterogeneity, rare cell populations, and complex regulatory networks that are obscured in bulk measurements [13].
Artificial Intelligence (AI) and Machine Learning (ML) have emerged as indispensable tools for extracting meaningful biological insights from this complexity. Their ability to identify subtle, non-linear patterns in large-scale data makes them particularly suited for tackling the analytical challenges inherent in modern transcriptomics, from managing data sparsity to integrating multi-omic datasets [95]. This technical guide explores the central role of AI and ML in advancing transcriptomic research, framed within the comparative context of bulk and single-cell methodologies.
Fundamental Differences Between Bulk and Single-Cell RNA-Seq
Understanding the application of AI requires a clear grasp of the fundamental technical and analytical differences between bulk and single-cell RNA-seq, as summarized in Table 1.
Table 1: Comparative Analysis of Bulk vs. Single-Cell RNA-Seq
| Feature | Bulk RNA-Seq | Single-Cell RNA-Seq |
|---|---|---|
| Resolution | Population-average [13] | Individual cell [13] |
| Key Applications | Differential gene expression, biomarker discovery, pathway analysis [13] | Cellular heterogeneity, rare cell identification, developmental trajectories, novel cell type discovery [13] [24] |
| Data Structure | Single expression vector per sample (gene x sample matrix) | High-dimensional matrix (gene x cell matrix) [95] |
| Primary Challenge | Masking of cell-specific signals [13] | Data sparsity, technical noise, computational scale [95] |
| Typical Workflow Complexity | Lower; established, straightforward pipelines [13] [96] | Higher; requires specialized steps for normalization, imputation, and clustering [13] [94] |
| Ideal AI/ML Approach | Supervised learning, regression models, classical statistical tests [97] | Unsupervised learning, deep learning for imputation, graph neural networks [98] [95] |
The experimental workflow for scRNA-seq introduces specific complexities not present in bulk protocols. Sample preparation requires the generation of viable single-cell suspensions, followed by meticulous quality control to ensure cell viability and integrity [13]. A crucial divergence is the cell partitioning step, where individual cells are isolated into micro-reaction vessels (e.g., GEMs) using microfluidic chips on specialized instruments [13]. This allows cell-specific barcoding, ensuring that transcripts can be traced back to their cell of origin—a foundational requirement for all subsequent single-cell analysis [13]. The following diagram outlines the core differential workflow and the resultant data structures that AI models must process.
Key AI and Machine Learning Applications in Transcriptomics
Dimensionality Reduction and Cell Type Identification
A primary challenge in scRNA-seq analysis is visualizing and interpreting data in thousands of dimensions. AI-driven dimensionality reduction techniques such as t-distributed Stochastic Neighbor Embedding (t-SNE) and Uniform Manifold Approximation and Projection (UMAP) are employed to project high-dimensional gene expression data into two or three dimensions for visualization and exploration [94]. Following this, unsupervised clustering algorithms (e.g., graph-based clustering, K-means) identify groups of cells with similar expression profiles, enabling the discovery of novel cell types and states [94]. These clusters are then annotated based on canonical marker genes, a process that can be accelerated by AI models trained on known cell type signatures [24].
Trajectory Inference and Lineage Reconstruction
Beyond identifying static cell types, scRNA-seq can reveal dynamic processes like differentiation. Trajectory inference methods use AI to reconstruct the sequence of transcriptional changes as cells transition from one state to another [24]. Tools like Monocle3 apply machine learning to order individual cells along a pseudotemporal trajectory based on their expression profiles, thereby reconstructing cellular differentiation pathways and lineage relationships without the need for time-series experiments [24].
Deep Learning for Data Imputation and Enhancement
The high sparsity of scRNA-seq data, due to technical dropouts (where a gene is expressed but not detected), is a major obstacle. Deep learning models are particularly effective for data imputation—distinguishing true biological zeros from technical zeros to recover missing gene expressions [98]. Models like gimVI and stPlus use variational autoencoders and other neural network architectures to leverage information from both the scRNA-seq data itself and complementary bulk or reference scRNA-seq datasets, enhancing data quality for downstream analysis [98].
Diagnostic and Prognostic Model Development
In clinical transcriptomics, AI excels at building predictive models from complex gene expression data. As demonstrated in a study on hypertrophic cardiomyopathy (HCM), researchers can systematically evaluate multiple machine-learning algorithms—such as XGBoost, Random Forest, and Support Vector Machines (SVM)—to construct robust diagnostic signatures from transcriptomic data [97]. This approach identified a compact 12-gene signature with superior diagnostic performance for HCM, showcasing the power of ML to distill complex molecular profiles into clinically actionable tools [97].
Multi-Omics and Spatial Data Integration
A frontier in transcriptomics is the integration of gene expression data with other molecular and spatial modalities. AI is critical for these data integration tasks. For instance, agentic AI systems like GenoMAS can automate the discovery of disease relationships by analyzing transcriptomic signatures across hundreds of disease-condition pairs, integrating multi-database enrichment analysis to quantify functional convergence [99]. Furthermore, with the rise of spatial transcriptomics (ST), novel computational approaches are required. Graph convolutional networks (e.g., SpaGCN) and multi-view autoencoders (e.g., MUSE) can integrate spatially resolved gene expression with histology images to identify spatial domains and cell-cell communication patterns, thereby preserving the critical spatial context of gene expression [98].
Table 2: AI/ML Techniques for Key Transcriptomic Analysis Tasks
| Analysis Task | AI/ML Method Category | Specific Tools/Models | Function |
|---|---|---|---|
| Dimensionality Reduction | Manifold Learning | UMAP, t-SNE [94] | Projects high-dim cell data to 2D/3D |
| Cell Clustering | Unsupervised Learning | Graph-based Clustering, K-means [94] | Identifies distinct cell populations |
| Trajectory Inference | Manifold Learning | Monocle3 [24] | Models cell differentiation paths |
| Data Imputation | Deep Learning | gimVI, stPlus, stLearn [98] | Corrects for technical noise/dropouts |
| Spatial Domain Detection | Graph Neural Networks | SpaGCN, MUSE [98] | Integrates gene expression with location |
| Diagnostic Modeling | Supervised Learning | XGBoost, Random Forest, SVM [97] | Builds predictive models from expression data |
| Disease Network Mapping | Agentic AI | GenoMAS [99] | Automates discovery of disease relationships |
Experimental Protocols and Workflows
Implementing AI in transcriptomic analysis requires a structured workflow, from raw data processing to biological interpretation. The following diagram and protocol outline a standard analysis pipeline for single-cell data, highlighting stages where AI/ML is critically applied.
Protocol: A Standard Workflow for AI-Driven scRNA-seq Analysis
Step 1: Raw Data Processing and Quality Control
- Processing: Raw sequencing reads (FASTQ files) are processed through pipelines like Cell Ranger to perform alignment, barcode assignment, and UMI counting, resulting in a feature-barcode matrix [94].
- Quality Control: Initial QC uses the pipeline's summary report (
web_summary.html). Subsequently, each cell is evaluated based on:- UMI Counts: Filtering out cells with unusually high (potential multiplets) or low (ambient RNA) counts.
- Genes Detected: Filtering cells with an extreme number of detected genes.
- Mitochondrial Read Percentage: Using a threshold (e.g., 10% for PBMCs) to remove low-quality or dying cells [94].
- AI/ML Role: Automated outlier detection and tools like SoupX or CellBender use ML models to estimate and subtract ambient RNA background [94].
Step 2: Normalization, Integration, and Feature Selection
- Normalization: Scaling methods are applied to correct for technical variation in sequencing depth between cells.
- Data Integration: If multiple samples/batches are analyzed, integration algorithms (e.g., Harmony) are used to remove batch effects while preserving biological variance [24].
- Feature Selection: Identifying highly variable genes that drive biological heterogeneity for downstream analysis.
Step 3: Core AI/ML Analysis
- Dimensionality Reduction: Apply UMAP or t-SNE to the highly variable genes to obtain a 2D/3D representation of the data [94].
- Clustering: Use graph-based clustering algorithms (e.g., Louvain/Leiden algorithm) to partition cells into distinct groups in the reduced space [94]. Annotate clusters using known marker genes.
- Differential Expression: Employ statistical models to find genes that are significantly differentially expressed between clusters or conditions. The
FindAllMarkersorFindMarkersfunction in Seurat is a common implementation [24]. - Advanced Analyses (as needed):
- Trajectory Inference: Input normalized expression matrices and cluster information into tools like Monocle3 to construct differentiation trajectories [24].
- Cell-Cell Communication: Use tools like CellChat to infer signaling interactions between cell clusters.
Step 4: Validation and Functional Interpretation
- Validation: Validate key findings using independent experimental methods (e.g., flow cytometry, immunohistochemistry) or external datasets [24] [97].
- Interpretation: Perform functional enrichment analysis (GO, KEGG) on lists of differentially expressed genes or genes specific to a trajectory using R packages like
clusterProfilerto derive biological meaning [24] [97].
The Scientist's Toolkit: Essential Research Reagents and Computational Tools
Successful implementation of the aforementioned workflows relies on a suite of wet-lab and computational tools.
Table 3: Essential Research Reagent Solutions and Computational Tools
| Item Name | Category | Function/Benefit |
|---|---|---|
| Chromium X Series | Instrument | Microfluidic instrument for single-cell partitioning into GEMs [13] [32] |
| GEM-X Flex / Universal Assays | Reagent Kit | Single-cell RNA-seq kits for barcoding and library prep; Flex enables high-throughput [13] |
| HISAT2 | Software | Splice-aware aligner for mapping RNA-seq reads to a reference genome [96] |
| Cell Ranger | Software Pipeline | Processes Chromium scRNA-seq data from FASTQ to feature-barcode matrices [94] |
| Seurat | R Toolkit | Comprehensive package for QC, normalization, clustering, and analysis of scRNA-seq data [24] |
| Loupe Browser | Visualization | Interactive desktop software for visual exploration and analysis of 10x Genomics data [94] |
| Monocle3 | R/Python Toolkit | Software for trajectory inference and analysis of single-cell expression data [24] |
| SpaGCN | Python Library | Graph convolutional network for integrating gene expression and spatial/histology data [98] |
The synergy between transcriptomic technologies and AI/ML is defining the next frontier of biological discovery and clinical application. As the field moves increasingly toward multi-omic integration at single-cell and spatial resolution, the data dimensionality and complexity will only intensify [98] [95]. The future lies in the development of more sophisticated, interpretable, and biologically grounded AI models—such as agentic AI systems and graph neural networks—that can not only scale with data size but also provide mechanistic insights into disease pathogenesis and treatment opportunities [99]. This progression will be crucial for realizing the full potential of precision medicine, transforming vast and complex transcriptomic datasets into actionable biological understanding and therapeutic breakthroughs.
Conclusion
Bulk and single-cell RNA-seq are not mutually exclusive technologies but rather complementary pillars of modern transcriptomics. Bulk RNA-seq remains a powerful, cost-effective tool for hypothesis generation and analyzing population-level changes in large cohorts. In contrast, single-cell RNA-seq is indispensable for dissecting cellular heterogeneity, discovering rare cell types, and understanding complex disease mechanisms at unprecedented resolution. The future lies in their strategic integration, supported by advanced computational methods like deconvolution and AI-driven analysis. This synergistic approach, combined with emerging multi-omics and spatial technologies, is rapidly accelerating biomarker discovery, drug development, and the advancement of precision medicine, ultimately leading to more targeted and effective therapies.
References
- 1. What is Bulk RNA sequencing (Bulk RNA-seq)? [https://www.scdiscoveries.com/support/what-is-bulk-rna-sequencing/]
- 2. From bulk, single-cell to spatial RNA sequencing [https://www.nature.com/articles/s41368-021-00146-0]
- 3. Bulk RNA Sequencing (RNA-seq) [https://science.nasa.gov/biological-physical/data/osdr/bulk-rna-sequencing-rna-seq/]
- 4. What is Bulk RNA Sequencing? [https://www.cd-genomics.com/resource-bulk-rna.html]
- 5. Perspectives on Bulk-Tissue RNA Sequencing and Single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11285642/]
- 6. [Tutorial] Bulk RNA-seq DE analysis [https://informatics.fas.harvard.edu/resources/tutorials/differential-expression-analysis/]
- 7. automated bulk RNA-seq exploration and visualisation using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8375142/]
- 8. Bulk RNA-seq Data Standards and Processing Pipeline [https://www.encodeproject.org/data-standards/rna-seq/long-rnas/]
- 9. Understanding Bulk and Single-Cell RNA Sequencing [https://www.cd-genomics.com/resource-bulk-rna-sequencing-vs-single-cell-rna-sequencing.html]
- 10. A practical guide to single-cell RNA-sequencing for ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0467-4]
- 11. Single‐cell RNA sequencing technologies and applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 12. Applications of single-cell RNA sequencing in drug ... [https://www.nature.com/articles/s41573-023-00688-4]
- 13. Bulk vs. single cell RNA-seq: Experimental differences and advantages ... [https://www.10xgenomics.com/blog/bulk-vs-single-cell-RNA-seq-experimental-differences-and-advantages-of-single-cell-resolution]
- 14. What Is The Difference Between Bulk Sequencing And Single-Cell ... [https://www.healthcarebusinesstoday.com/what-is-the-difference-between-bulk-sequencing-and-single-cell-sequencing/]
- 15. Single cell RNA-seq: An introductory overview and tools for ... [https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started]
- 16. Single Cell Sequencing and The Future of Drug Discovery [https://www.parsebiosciences.com/blog/single-cell-sequencing-and-the-future-of-drug-discovery/]
- 17. Single-cell technology for drug discovery and development [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2024.1459962/full]
- 18. Should I Choose Single Cell Sequencing or Bulk ... [https://singleron.bio/should-i-choose-single-cell-sequencing-or-bulk-sequencing/]
- 19. Challenges and Considerations in Bulk RNA-seq [https://alitheagenomics.com/blog/challenges-and-considerations-in-bulk-rna-seq]
- 20. Clinical application of single‐cell RNA sequencing in disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576031/]
- 21. Single-Cell RNA-Seq Workshop 2025 [https://www.ecseq.com/workshops/workshop_2025-08-Single-Cell-RNA-Seq-Data-Analysis]
- 22. Global trends in machine learning applications for single-cell ... [https://hereditasjournal.biomedcentral.com/articles/10.1186/s41065-025-00528-y]
- 23. Comprehensive analysis of single-cell and bulk RNA ... [https://www.nature.com/articles/s41598-025-23779-1]
- 24. Integrated analysis of single-cell RNA-seq and bulk RNA-seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548165/]
- 25. Progress and applications of single-cell RNA sequencing ... [https://www.sciencedirect.com/science/article/pii/S2162253125001374]
- 26. What is the difference between bulk and single-cell RNA ... [https://synapse.patsnap.com/article/what-is-the-difference-between-bulk-and-single-cell-rna-seq-analysis]
- 27. New review decodes transcriptomics: bulk vs. single-cell ... [https://www.eurekalert.org/news-releases/1103818]
- 28. Whole Transcriptome vs 3' mRNA-Seq: How to Choose ... [https://www.lexogen.com/blog/choosing-between-whole-transcriptome-total-rna-seq-and-3-mrna-seq/]
- 29. Whole Transcriptome vs. Targeted Gene Expression Profiling [https://missionbio.com/company/blog/comparison-whole-transcriptome-vs-targeted-gene-expression-profiling/]
- 30. Optimized murine sample sizes for RNA sequencing ... [https://www.nature.com/articles/s41467-025-65022-5]
- 31. Differential detection workflows for multi-sample single-cell ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12102-x]
- 32. Comparison of single-cell RNA-seq methods to enable ... [https://www.sciencedirect.com/science/article/pii/S2667237525002097]
- 33. Bioinformatics perspectives on transcriptomics [https://www.rna-seqblog.com/bioinformatics-perspectives-on-transcriptomics-a-comprehensive-review-of-bulk-and-single-cell-rna-sequencing-analyses/]
- 34. Bulk RNA Sequencing versus Single-Cell RNAseq: 2 Powerful... [https://sampled.com/bulk-rna-sequencing-vs-single-cell-rna-sequencing/]
- 35. Applications of single-cell RNA sequencing in drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141847/]
- 36. Deep transfer learning of cancer drug responses by ... [https://www.nature.com/articles/s41467-022-34277-7]
- 37. Functional phenotyping of genomic variants using joint ... [https://www.nature.com/articles/s41592-025-02805-0]
- 38. The Beginner's guide to bulk RNA-Seq Analysis [https://silicogene.com/blog/bulk-rna-seq-data-analysis/]
- 39. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 40. Statistical and Bioinformatics Analysis of Data from Bulk and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7771369/]
- 41. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 42. Advancements in single-cell RNA sequencing and spatial ... [https://pubmed.ncbi.nlm.nih.gov/39980637/]
- 43. Navigating single-cell RNA-sequencing: protocols, tools ... [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00044-5]
- 44. Global trends in machine learning applications for single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12357469/]
- 45. Deep generative modeling of sample-level heterogeneity ... [https://www.nature.com/articles/s41592-025-02808-x]
- 46. Integrating bulk and single cell RNA-seq refines ... [https://elifesciences.org/reviewed-preprints/106183]
- 47. Integration of Single-cell and bulk RNA sequencing data ... [https://www.nature.com/articles/s41598-025-22803-8]
- 48. RNA Sequencing in Drug Discovery and Development [https://www.lexogen.com/blog/rna-sequencing-in-drug-discovery-and-development/]
- 49. Current best practices in single‐cell RNA‐seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 50. 16. Differential gene expression analysis [https://www.sc-best-practices.org/conditions/differential_gene_expression.html]
- 51. Integrated single-cell and bulk RNA dequencing to identify ... [https://pubmed.ncbi.nlm.nih.gov/40635996/]
- 52. Identification of leukemia-enriched signature through the ... [https://www.nature.com/articles/s41467-025-59362-5]
- 53. AACR 2025: Single-cell RNA sequencing provides ... [https://oncologynews.com.au/latest-news/aacr-2025-single-cell-rna-sequencing-provides-comprehensive-map-of-acute-myeloid-leukemia-cell-states/]
- 54. Application and research progress of single cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11390353/]
- 55. Single cell RNA sequencing improves the next generation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11783831/]
- 56. A multiplex single-cell RNA-Seq pharmacotranscriptomics ... [https://www.nature.com/articles/s41589-024-01761-8]
- 57. Exploring and mitigating shortcomings in single-cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03525-6]
- 58. Replicability of bulk RNA-Seq differential expression and ... [https://pubmed.ncbi.nlm.nih.gov/40324149/]
- 59. Functional phenotyping of genomic variants using joint ... [https://pubmed.ncbi.nlm.nih.gov/40890552/]
- 60. Single-cell Analysis Market to Reach USD 12.29 Billion by ... [https://finance.yahoo.com/news/single-cell-analysis-market-reach-085100637.html]
- 61. Single Cell Transcriptome Sequencing: Costs and ... [https://www.scdiscoveries.com/blog/knowledge/single-cell-transcriptome-sequencing-costs-considerations/]
- 62. High-Throughput Single Cell Sequencing Platform 2025-2033 ... [https://www.archivemarketresearch.com/reports/high-throughput-single-cell-sequencing-platform-771854]
- 63. Enabling scalable single-cell transcriptomic analysis ... [https://www.nature.com/articles/s41598-025-12897-5]
- 64. ScaleSC: a superfast and scalable single-cell RNA-seq data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12321287/]
- 65. Single Cell Sequencing Market Size 2025 to 2034 [https://www.precedenceresearch.com/single-cell-sequencing-market]
- 66. Advancing Single-Cell RNA-Seq: Integration & Multi-Omics [https://www.elucidata.io/blog/advancing-single-cell-rna-seq-analysis]
- 67. DropDAE: Denosing Autoencoder with Contrastive Learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383624/]
- 68. SinCWIm: An imputation method for single-cell RNA ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482524003093]
- 69. New Single cell Methods Guide 2025 [https://www.rna-seqblog.com/new-single-cell-methods-guide-2025/]
- 70. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 71. Robustness and resilience of computational deconvolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159287/]
- 72. RNA-Seq Normalization: Methods and Stages [https://bigomics.ch/blog/why-how-normalize-rna-seq-data/]
- 73. A benchmark of RNA-seq data normalization methods for ... [https://www.nature.com/articles/s41540-024-00448-z]
- 74. Benchmark of cellular deconvolution methods using a multi ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03552-3]
- 75. Navigating single-cell RNA-sequencing: protocols, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 76. Single-cell RNA sequencing reveals different cellular ... [https://www.nature.com/articles/s41523-025-00808-w]
- 77. 10 Best Practices for Effective RNA-Seq Data Analysis - Focal [https://www.getfocal.co/post/10-best-practices-for-effective-rna-seq-data-analysis]
- 78. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 79. Transcriptome size matters for single-cell RNA-seq ... [https://www.nature.com/articles/s41467-025-56623-1]
- 80. St. Jude researchers create tool incorporating ... [https://www.stjude.org/media-resources/news-releases/2025-medicine-science-news/st-jude-researchers-create-tool-incorporating-transcriptome-size-to-improve-rna-seq-analysis.html]
- 81. A distribution-independent cell deconvolution model for ... [https://www.sciencedirect.com/science/article/pii/S2666979X2500206X]
- 82. New generative methods for single-cell transcriptome data ... [https://www.nature.com/articles/s41598-024-54798-z]
- 83. sNucConv: A bulk RNA-seq deconvolution method trained ... [https://www.sciencedirect.com/science/article/pii/S2589004224015931]
- 84. From pixels to cell types: a comprehensive review of ... [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00055-2]
- 85. Protocol for interpretable and context-specific single-cell- ... [https://www.sciencedirect.com/science/article/pii/S2666166725000760]
- 86. Comparative Analysis of Single-Cell RNA Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11011421/]
- 87. Droplet-based single-cell RNA sequencing: decoding cellular ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06996-0]
- 88. Concepts and new developments in droplet-based single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11568944/]
- 89. The Single Cell Solution Immunology Has Been... - Parse Biosciences [https://www.parsebiosciences.com/blog/the-single-cell-solution-immunology-has-been-waiting-for/]
- 90. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 91. UDA-seq: universal droplet microfluidics-based ... [https://www.nature.com/articles/s41592-024-02586-y]
- 92. Applications and emerging challenges of single-cell RNA ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000030]
- 93. Spatial integration of multi-omics single-cell data with SIMO [https://www.nature.com/articles/s41467-025-56523-4]
- 94. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 95. Eleven grand challenges in single-cell data science [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-1926-6]
- 96. Introduction to RNA-seq Analysis - SCINet - USDA [https://scinet.usda.gov/events/2025-05-13-rna-seq]
- 97. Frontiers | Transcriptomic and machine learning modeling... analysis [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1596049/full]
- 98. Computational Approaches and Challenges in Spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10372921/]
- 99. [2508.04742] Discovery of Disease Relationships via Transcriptomic ... [https://arxiv.org/abs/2508.04742]