Mastering RBP-RNA Interactions: A Comprehensive Guide to CLIP-Seq Protocols (HITS-CLIP, PAR-CLIP, iCLIP)
This article provides a detailed, comparative analysis of the major crosslinking and immunoprecipitation (CLIP-seq) protocols—HITS-CLIP, PAR-CLIP, and iCLIP—for mapping RNA-binding protein (RBP) interactions.
Mastering RBP-RNA Interactions: A Comprehensive Guide to CLIP-Seq Protocols (HITS-CLIP, PAR-CLIP, iCLIP)
Abstract
This article provides a detailed, comparative analysis of the major crosslinking and immunoprecipitation (CLIP-seq) protocols—HITS-CLIP, PAR-CLIP, and iCLIP—for mapping RNA-binding protein (RBP) interactions. Targeted at researchers and drug developers, it covers the foundational principles of each method, step-by-step application workflows, common troubleshooting and optimization strategies, and a critical validation framework for data analysis. The guide aims to empower scientists to select and implement the optimal CLIP protocol for their specific RBP of interest, advancing the study of post-transcriptional regulation in health and disease.
Decoding the CLIP-Seq Universe: Foundational Principles of HITS-CLIP, PAR-CLIP, and iCLIP
Protein-RNA interactions (PRIs) form the operational bedrock of post-transcriptional gene regulation. Mapping these interactions is a core objective in molecular biology because it directly deciphers the regulatory code controlling RNA fate—including its splicing, stability, localization, and translation. Dysregulation of these interactions by RNA-binding proteins (RBPs) is a fundamental mechanism underlying numerous diseases, including neurodegenerative disorders (e.g., ALS, Alzheimer's), cancer, and autoimmune conditions. The thesis of this document is that CLIP-seq derivative protocols (HITS-CLIP, PAR-CLIP, iCLIP) are indispensable tools for in vivo PRI mapping, each offering unique advantages for elucidating mechanistic insights into gene regulation and identifying novel therapeutic targets.
Quantitative Comparison of CLIP-Seq Methodologies
The evolution of CLIP-seq protocols has addressed specific technical challenges, leading to varied applications. Key quantitative metrics are summarized below.
Table 1: Comparative Analysis of Major CLIP-Seq Protocols
| Protocol | Crosslinking Method | Key Mutational/Truncation Signature | Primary Resolution | Key Advantage | Common RBP Applications |
|---|---|---|---|---|---|
| HITS-CLIP | UV-C (254 nm) | Deletions (RNA truncation at crosslink site) | ~30-60 nt (binding region) | Robust, widely applicable; defines binding regions. | Splicing regulators (e.g., NOVA, RBFOX), miRNAs. |
| PAR-CLIP | UV-A (365 nm) + 4-Thiouridine/6-Thioguanosine | T-to-C (4SU) or G-to-A (6SG) transitions | Single-nucleotide (when high mutation rate) | Highest crosslinking efficiency & nucleotide-resolution mapping. | Detailed mechanistic studies of RBP binding motifs. |
| iCLIP | UV-C (254 nm) | cDNA truncation at crosslink site (+1 position) | Single-nucleotide (via truncation site) | Maps exact crosslink site; captures transient interactions. | Complex RBPs (e.g., TDP-43, FUS), splicing analysis. |
| eCLIP | UV-C (254 nm) | Size-matched input controls, improved specificity | ~30-60 nt | Reduced artifact signals; ENCODE standard. | Systematic profiling (ENCODE projects). |
Experimental Protocols
Core CLIP-Seq Workflow
This universal framework underpins all variant protocols.
Protocol: Core CLIP-Seq Experimental Steps
- In Vivo Crosslinking: Live cells or tissue are subjected to UV irradiation (254 nm for protein-nucleic acid crosslinking).
- Cell Lysis and Fragmentation: Cells are lysed in stringent buffer. RNA is partially fragmented by limited RNase digestion.
- Immunoprecipitation (IP): Lysate is incubated with antibodies specific to the RBP of interest, coupled to magnetic beads.
- Stringent Washes: Beads are washed with high-salt buffers to remove non-specific associations.
- RNA Linker Ligation: A 3' RNA adapter is ligated to the RNA fragments bound to the RBP.
- Radiolabeling & Transfer (Optional): For visualization, RNA-protein complexes are radiolabeled, separated by SDS-PAGE, and transferred to a nitrocellulose membrane.
- Proteinase K Digestion & RNA Extraction: Protein is digested, and crosslinked RNA is purified.
- Reverse Transcription & cDNA Ligation: RNA is reverse transcribed. A cDNA adapter is ligated to the 3' end of the cDNA.
- PCR Amplification & Sequencing: Libraries are amplified and sequenced using high-throughput platforms.
Protocol Highlight: iCLIP for Precise Crosslink Mapping
iCLIP introduces a critical modification during reverse transcription.
Protocol: Key iCLIP-Specific Steps
- Follow core CLIP steps 1-5.
- After membrane transfer and proteinase K digestion, extract RNA.
- Reverse Transcription with Premature Termination: Use reverse transcriptase that tends to terminate at the crosslinked nucleotide (+1 position of cDNA).
- cDNA Circularization: Instead of blunt-end ligation, the cDNA is circularized using Circligase.
- PCR Amplification: Use primers designed to linearize the circular cDNA and add sequencing adapters.
- Bioinformatic Analysis: Map the truncation sites in sequencing reads to identify the exact crosslink nucleotide.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CLIP-Seq Experiments
| Item | Function & Importance | Example/Note |
|---|---|---|
| UV Crosslinker | Induces covalent bonds between RBPs and RNA in vivo. | UV-C (254 nm) for HITS/iCLIP; UV-A (365 nm) for PAR-CLIP. |
| RNase I | Partially digests RNA to generate bound fragments of optimal length. | Concentration is titrated for each RBP. |
| Magnetic Protein A/G Beads | Solid support for antibody-based immunoprecipitation. | Enable stringent washing. |
| High-Affinity RBP Antibody | Specifically captures the RBP-RNA complex. | Validation for IP is critical; FLAG/HA tags can be used. |
| T4 RNA Ligase 1 (truncated) | Ligates RNA adapter to 3' end of fragmented RNA. | Works on RNA with 3'-OH (created by RNase fragmentation). |
| Proteinase K | Digests the RBP to release crosslinked RNA for library prep. | Must be molecular biology grade, RNase-free. |
| Reverse Transcriptase (Superscript III/IV) | Synthesizes cDNA from crosslinked RNA template. | For iCLIP, uses conditions promoting truncation at crosslink site. |
| Circligase ssDNA Ligase | Circularizes cDNA in iCLIP protocol. | Enables capture of truncated cDNAs. |
| 4-Thiouridine (4SU) | Photosensitive nucleoside analog incorporated into RNA for PAR-CLIP. | Increases crosslinking efficiency and induces T-to-C mutations. |
Visualizations
CLIP-Seq Core Experimental Workflow
RBP Dysregulation Leads to Disease
Protocol Choice Dictates Application
Within the broader thesis on CLIP-Seq methodologies (HITS-CLIP, PAR-CLIP, iCLIP), this document details the fundamental application of ultraviolet (UV) crosslinking to capture transient, native interactions between RNA-binding proteins (RBPs) and their RNA targets. This paradigm is the critical first step that enables high-resolution mapping of RBP binding sites across the transcriptome, informing basic molecular biology and drug development for RNA-centric therapies.
Application Notes
Core Principle of UV Crosslinking
UV light at 254 nm induces the formation of covalent bonds between RBPs and RNAs that are in direct, intimate contact at the moment of irradiation. This "freezes" otherwise transient complexes, allowing for stringent purification that removes non-specifically associated RNAs.
Key Advantages:
- Snapshot of Native State: Captures interactions in vivo or in physiologic conditions in vitro.
- High Specificity: Covalent linkage permits rigorous washing, dramatically reducing background noise.
- Compatibility: Forms the basis for all subsequent CLIP variant protocols.
Quantitative Performance Metrics: The efficiency of crosslinking is a critical parameter influencing downstream success.
Table 1: UV Crosslinking Parameters and Outcomes
| Parameter | Typical Range / Value | Impact / Note |
|---|---|---|
| UV Wavelength | 254 nm | Optimal for creating protein-nucleic acid crosslinks. |
| Energy Delivery | 150-400 mJ/cm² (in vivo) | Varies by cell type, tissue depth, and RBP. Must be optimized to balance crosslink yield with cellular damage. |
| Crosslinking Efficiency | 1-5% of a given RBP-RNA complex | Inherently low, but sufficient for library preparation due to PCR amplification. |
| Cell Viability Post-UV | 50-80% (at 150-200 mJ/cm²) | Must be monitored to ensure representative sampling. |
| RNA Fragment Size Post-RNase | 20-60 nucleotides | Defines the resolution of binding site mapping. |
Protocol: In Vivo UV Crosslinking of Adherent Cells
This protocol describes the foundational step for most CLIP-seq experiments.
Materials & Reagents:
- PBS (ice-cold)
- UV Crosslinker (254 nm, e.g., Stratagene Stratalinker)
- Cell scraper
- Liquid nitrogen
- Lysis Buffer (e.g., 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease inhibitors, RNase inhibitors)
Methodology:
- Culture & Preparation: Grow adherent cells to ~80-90% confluency in 150 mm plates.
- Wash: Aspirate medium and wash cells twice gently with ice-cold PBS.
- UV Irradiation: Remove PBS completely. Place open plate on ice. Irradiate cells with 254 nm UV light at 150-200 mJ/cm². An un-irradiated control plate should be processed in parallel.
- Harvest: Immediately after irradiation, add 1 mL ice-cold PBS, scrape cells, and transfer to a pre-chilled microcentrifuge tube.
- Pellet: Centrifuge at 500 x g for 3 min at 4°C. Aspirate supernatant.
- Flash-Freeze: Snap-freeze cell pellet in liquid nitrogen. Store at -80°C or proceed to lysis.
- Lysis: Resuspend pellet in 1 mL of strong lysis buffer with fresh inhibitors. Vortex vigorously. Incubate on ice for 15 min.
- Clarify: Centrifuge at 16,000 x g for 15 min at 4°C. Transfer supernatant (the lysate containing crosslinked complexes) to a new tube. Lysate is now ready for immunoprecipitation.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for CLIP-Seq Crosslinking & Isolation
| Item | Function & Importance |
|---|---|
| 254 nm UV Crosslinker | Precise, calibrated delivery of UV energy for consistent covalent crosslinking. |
| RNase Inhibitors (e.g., RiboLock) | Critical to prevent degradation of RNA targets during cell lysis and processing post-UV. |
| Complete Protease Inhibitor Cocktail | Preserves the RBP and its crosslinked RNA adduct during lysis. |
| Paramagnetic Protein A/G Beads | For efficient immunoprecipitation of the RBP-RNA complex; enable stringent washing. |
| Sequence-Specific RNase (e.g., RNase T1, RNase I) | Fragments RNA to isolate protein-protected regions; choice defines binding site resolution. |
| Phosphatase & Kinase Enzymes | Used in iCLIP to prepare RNA ends for adapter ligation (dephosphorylation) and in PAR-CLIP for nucleoside analog incorporation. |
| 4-Thiouridine (4-SU) or 6-Thioguanosine (6-SG) | Photosensitive nucleoside analogs for PAR-CLIP; increase crosslinking efficiency and induce diagnostic T>C mutations. |
| T4 PNK (Polynucleotide Kinase) | Essential for radio-labeling RNA 5' ends (for visualization) and for 3' phosphatase/5' kinase reactions in iCLIP. |
| TruSeq or NEXTflex CLIP-seq Adapters | Specialized adapters for ligation to fragmented, protein-bound RNA with minimal bias. |
Visualization of the CLIP-Seq Paradigm
Diagram 1: The CLIP-Seq Experimental Workflow
Diagram 2: Logic of UV Crosslinking for RBP Studies
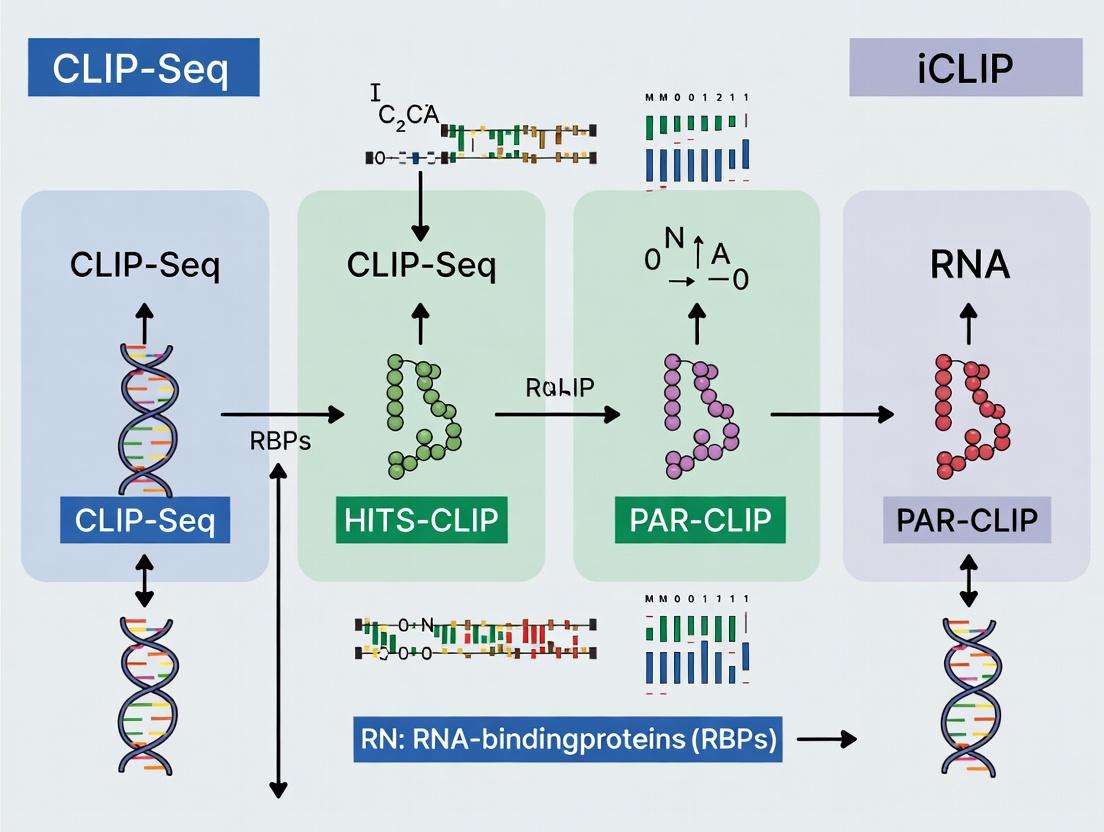
Historical Context and Evolution within CLIP-Seq Methodologies
The development of CLIP-seq (Crosslinking and Immunoprecipitation followed by sequencing) methodologies represents a pivotal advancement in the study of RNA-protein interactions. UV crosslinking, specifically using UV-C light at 254 nm, is the foundational step that enables the covalent fixation of protein to RNA at zero-distance interaction sites. This principle, established in the 1990s, was first scaled genome-wide in the landmark 2005 study by Ule et al., which introduced HITS-CLIP (High-Throughput Sequencing of RNA isolated by CLIP). This protocol solved the critical problem of identifying in vivo RNA binding protein (RBP) binding sites at nucleotide resolution, providing a direct snapshot of the RNA interactome.
The standard UV-C crosslinking protocol became the benchmark against which subsequent variations were developed. PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced CLIP), introduced in 2010, incorporates nucleoside analogs (e.g., 4-thiouridine) and uses 365 nm UVA light, inducing T-to-C transitions in sequencing reads for higher confidence mapping. iCLIP (individual-nucleotide resolution CLIP), also developed in 2010, introduced a circularization step to capture the cDNA of the crosslinked RNA fragment, allowing for the precise identification of crosslink sites and the study of truncated cDNAs. The historical trajectory from HITS-CLIP to these more recent methods is defined by iterative improvements in crosslinking efficiency, background reduction, and mapping precision, all within the broader thesis of deciphering post-transcriptional regulatory networks controlled by RBPs.
Quantitative Comparison of Key CLIP-Seq Variants
The table below summarizes the core quantitative parameters that differentiate the major CLIP-seq protocols, centered on their crosslinking approach.
Table 1: Quantitative Comparison of CLIP-Seq Methodologies
| Parameter | HITS-CLIP (Standard) | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslink Type | UV-C (254 nm) | UVA (365 nm) with 4-thiouridine (4SU) | UV-C (254 nm) |
| Crosslink Efficiency | ~1-5% (depends on RBP-RNA interface) | ~5-20% (enhanced by 4SU) | ~1-5% (similar to HITS-CLIP) |
| Characteristic Mutation | None (but can have deletions at crosslink site) | T-to-C (from 4SU) or G-to-A (from 6SG) | Truncated cDNAs at crosslink site |
| Typical Resolution | ~30-60 nucleotides | Single-nucleotide (via mutation mapping) | Single-nucleotide (via cDNA truncation) |
| Key Diagnostic Read | Crosslink-induced deletions in reads | Non-physiological mutation rate in peaks | cDNA truncation site (start of read) |
| Primary Advantage | Robust, works in vivo, no metabolic labeling required | Highest signal-to-noise, precise site identification | Identifies exact crosslink site, studies truncations |
| Primary Limitation | Lower crosslinking efficiency, harder to map precise site | Requires metabolic labeling, may perturb cell physiology | More complex library prep, lower yield |
Detailed Protocol: Standard UV-C HITS-CLIP
This protocol is designed for cultured mammalian cells.
Part A: In Vivo UV-C Crosslinking and Cell Lysis
- Materials: Adherent cells at ~90% confluence, Ice-cold PBS, UV-C Crosslinker (254 nm, e.g., Stratagene Stratalinker), Lysis Buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate, supplemented with RNase Inhibitor and protease inhibitors).
- Procedure:
- Aspirate culture medium and wash cells twice with 10 mL of ice-cold PBS.
- Aspirate PBS completely. Place culture dish on ice.
- UV Irradiation: In a pre-chilled UV-C crosslinker, irradiate cells at 254 nm with 200-400 mJ/cm². Optimization Note: Energy must be titrated for each RBP to balance crosslinking efficiency versus RNA degradation.
- Immediately after irradiation, add 1 mL of lysis buffer per 10 cm dish. Scrape cells and transfer lysate to a microcentrifuge tube.
- Sonicate lysate briefly (3 x 5 sec pulses at low power) to shear DNA and reduce viscosity. Clarify by centrifuging at 20,000 x g for 10 min at 4°C. Transfer supernatant to a new tube.
Part B: Partial RNase Digestion and Immunoprecipitation
- Materials: RNase I (Ambion), Dynabeads Protein A/G, Target-specific antibody (validated for CLIP), High-Salt Wash Buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate), Low-Salt Wash Buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl₂, 0.2% Tween-20).
- Procedure:
- To the clarified lysate, add RNase I to a final dilution of 1:1000 to 1:50,000. Incubate at 22°C for 5-15 min. This is a critical optimization step to generate ~50-100 nt RNA fragments bound by the RBP.
- Pre-clear lysate with 20 µL of bare beads for 30 min at 4°C.
- Couple 5 µg of specific antibody (or IgG control) to 50 µL of Protein A/G beads for 1 hour at room temperature.
- Incubate the pre-cleared, RNase-treated lysate with antibody-bound beads for 2 hours at 4°C with rotation.
- Wash beads sequentially: 2x with High-Salt Wash Buffer, 1x with Low-Salt Wash Buffer.
Part C: RNA Adapter Ligation, Radiolabeling, and Isolation
- Materials: T4 PNK (3' phosphatase minus), [γ-³²P] ATP, T4 RNA Ligase 1, Pre-adenylated 3' adapter, 5' RNA adapter, 10% Tris-Glycine SDS-PAGE gel, Nitrocellulose membrane.
- Procedure:
- On-bead 3' Dephosphorylation: Use T4 PNK (3' phosphatase minus) in 1x PNK buffer for 20 min at 37°C. Wash beads.
- 3' Adapter Ligation: Ligate pre-adenylated 3' adapter using T4 RNA Ligase 1 (truncated) in ligation buffer overnight at 16°C. Wash.
- 5' End Radiolabeling: Use T4 PNK and [γ-³²P] ATP in 1x PNK buffer for 20 min at 37°C. This labels the RNA 5' ends created by RNase cleavage. Wash thoroughly.
- Complex Elution and Transfer: Elute RBP-RNA complexes by boiling beads in 1x SDS loading buffer. Run eluate on a 10% Tris-Glycine SDS-PAGE gel.
- Membrane Transfer and Excision: Transfer proteins to a nitrocellulose membrane. Expose membrane to a phosphorimager screen for 1-2 hours. Excise the membrane region corresponding to the full-length RBP (shifted up by ~20-30 kDa due to crosslinked RNA).
Part D: Proteinase K Treatment, RNA Extraction, and Library Prep
- Materials: Proteinase K, 7 M Urea, Acid-phenol:chloroform, Glycogen, Reverse transcription primers, High-fidelity PCR polymerase.
- Procedure:
- Incubate excised membrane pieces in Proteinase K buffer with 1% SDS for 20 min at 37°C, then 20 min at 55°C.
- Extract RNA by adding 7 M urea and acid-phenol:chloroform. Precipitate with glycogen.
- Reverse transcribe the RNA using a primer complementary to the 3' adapter.
- Amplify cDNA by PCR (12-18 cycles) using primers containing Illumina flow cell adapters and sample barcodes.
- Purify PCR product via gel electrophoresis or solid-phase reversible immobilization (SPRI) beads. Validate library quality via Bioanalyzer and quantify by qPCR before sequencing.
Visualizations
Diagram 1: HITS-CLIP Experimental Workflow
Diagram 2: Historical Evolution of CLIP-Seq Methods
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Standard UV-C HITS-CLIP
| Reagent / Material | Function / Role in Protocol | Key Considerations |
|---|---|---|
| UV-C Crosslinker (254 nm) | Induces covalent bonds between RBP and directly contacting RNA nucleotides at zero distance. | Must deliver calibrated, reproducible energy (mJ/cm²). Cooled stage minimizes heat denaturation. |
| RNase I (Partial Digestion Grade) | Trims unprotected RNA to leave ~50-100 nt fragments protected by the bound RBP, defining binding site resolution. | Concentration is critical; must be titrated for each RBP to avoid over-digestion. |
| Magnetic Beads (Protein A/G) | Solid-phase support for immunoprecipitation of the RBP-RNA complex via a specific antibody. | Provide low non-specific RNA binding and efficient washing. |
| Validated RBP-Specific Antibody | Provides specificity for immunoprecipitating the target RBP-RNA complex from the lysate. | Must recognize native, crosslinked protein. CLIP-validated antibodies are preferred. |
| Pre-adenylated 3' Adapter | Modified adapter ligated to the 3' end of the RNA fragment using a truncated ligase, preventing adapter self-ligation. | Essential for efficient library construction from the small amount of recovered RNA. |
| [γ-³²P] ATP & T4 PNK | Radiolabels the 5' phosphate of the RNA fragment post-RNase cleavage, enabling visualization on a membrane after SDS-PAGE. | Allows precise excision of the RBP-RNA complex region, reducing background from free protein or RNA. |
| Nitrocellulose Membrane | Binds proteins during transfer from SDS-PAGE gel. Retains the covalently linked RNA-protein complex. | PVDF is not suitable as it does not retain RNA. |
| Proteinase K | Digests the RBP after membrane excision, releasing the crosslinked RNA fragment for purification and library prep. | Must be molecular biology grade, free of RNases. |
Within the landscape of CLIP-seq methodologies—including HITS-CLIP, iCLIP, and others—Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) stands out for its precision in defining RNA-protein interaction sites. Its core innovation is the incorporation of the nucleoside analog 4-thiouridine (4SU) into nascent RNA, which, upon UV crosslinking at 365 nm, generates characteristic T-to-C transitions in cDNA sequences. This signature provides nucleotide-resolution mapping of RNA-binding protein (RBP) footprints, reducing background and enabling highly accurate binding site identification—a critical advantage for research in post-transcriptional regulation and drug target discovery.
Table 1: Comparison of Key CLIP-seq Methodologies
| Feature | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslink Agent | UV-C (254 nm) | UV-A (365 nm) + 4SU | UV-C (254 nm) |
| Characteristic Mutation | Deletions, truncations | T-to-C transitions | cDNA truncations at crosslink site |
| Signal-to-Noise | Moderate | High (due to mutation signature) | High |
| Binding Resolution | ~30-60 nt | Nucleotide-level | Nucleotide-level |
| Key Requirement | High antibody specificity | 4SU incorporation efficiency | Specialized adapter for truncation |
| Primary Output | Crosslink-induced mutation sites (CIMS) | Transition sites (T-to-C) | Crosslink-induced truncation sites (CITS) |
Table 2: Typical Experimental Parameters and Outcomes for PAR-CLIP
| Parameter | Typical Range / Value | Notes |
|---|---|---|
| 4SU Concentration | 100 - 500 µM | Cell type-dependent; non-toxic dose. |
| 4SU Incubation Time | 12 - 16 hours | Ensures sufficient incorporation. |
| Crosslink Wavelength | 365 nm | Optimized for 4SU reactivity. |
| Crosslink Energy | 0.15 - 0.30 J/cm² | Typically delivered by a UV-A lamp. |
| T-to-C Transition Rate | 2 - 20% at binding site | >2% is indicative of true crosslink. |
| Sequencing Depth | 10 - 30 million reads | Sufficient for robust site identification. |
Detailed PAR-CLIP Protocol
Cell Culture and 4SU Incorporation
- Culture cells (e.g., HEK293) in standard medium.
- Supplement medium with 100-500 µM 4-thiouridine (4SU). Use a DMSO vehicle control for a parallel "no-4SU" experiment.
- Incubate for 12-16 hours under normal growth conditions to allow 4SU incorporation into nascent RNA.
In Vivo Crosslinking & Cell Lysis
- Wash cells twice with ice-cold PBS.
- Crosslink using UV light at 365 nm (0.15-0.30 J/cm²) on ice in PBS. For adherent cells, perform this in the culture dish.
- Harvest cells by scraping and pellet by centrifugation.
- Lyse cells in strong lysis buffer (e.g., 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- Shear DNA by brief sonication or passage through a fine-gauge needle. Clarify lysate by centrifugation.
Immunoprecipitation and RNA Handling
- Pre-clear lysate with protein A/G beads for 30 min at 4°C.
- Incubate supernatant with antibody against the target RBP, conjugated to magnetic beads, for 1-2 hours at 4°C.
- Wash beads stringently with high-salt and detergent buffers to remove non-specific interactions.
- Dephosphorylate 3' RNA ends on beads using Antarctic Phosphatase.
- Ligate a pre-adenylated 3' adapter using T4 RNA Ligase 1 (truncated).
- Radio-label 5' RNA ends with [γ-³²P]ATP and T4 PNK.
- Resolve RNP complexes by SDS-PAGE. Transfer to a nitrocellulose membrane.
- Expose membrane to a phosphor screen, excise the band corresponding to the RBP-RNA complex.
- Proteinase K digest the excised band to recover crosslinked RNA.
Library Construction and Sequencing
- Reverse transcribe the RNA using a primer complementary to the 3' adapter. This step is where T-to-C mutations are introduced during cDNA synthesis opposite the crosslinked 4SU.
- Ligate the 5' adapter to the cDNA.
- PCR amplify the libraries using indexing primers.
- Sequence on an Illumina platform, focusing on single-end, 50-75 bp reads.
Data Analysis Core
- Align reads to the genome using aligners tolerant of mismatches (e.g., Bowtie, STAR).
- Identify T-to-C transitions in aligned reads. Cluster transition sites to identify significant peaks.
- Filter peaks based on the transition rate (T-to-C / (T-to-C + T)) to distinguish true binding sites from background.
Visualizations
Title: PAR-CLIP Experimental Workflow
Title: Molecular Basis of T-to-C Signature in PAR-CLIP
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PAR-CLIP Experiments
| Reagent / Material | Function / Purpose | Critical Notes |
|---|---|---|
| 4-Thiouridine (4SU) | Nucleoside analog incorporated into RNA; photosensitizer for efficient 365 nm crosslinking. | Optimize concentration for cell type to minimize toxicity. |
| UV-A Lamp (365 nm) | Light source for specific crosslinking of 4SU to RBPs. | Calibrate energy output (J/cm²) for reproducible crosslinking. |
| High-Affinity RBP Antibody | For specific immunoprecipitation of the target RNP complex. | Specificity is paramount; validate for IP. |
| Protein A/G Magnetic Beads | Solid support for antibody-mediated capture of complexes. | Enable efficient washing and buffer exchange. |
| Pre-adenylated 3' Adapter | Ligated to RNA 3' ends after IP; prevents adapter dimer formation. | Essential for ligation without ATP. |
| T4 RNA Ligase 1 (truncated K227Q) | Ligates the pre-adenylated adapter to RNA. | Minimizes side reactions. |
| Proteinase K | Digests the RBP after membrane excision to recover crosslinked RNA. | Must be molecular biology grade, RNase-free. |
| Reverse Transcriptase | Synthesizes cDNA from crosslinked RNA; enzyme properties influence mutation signature. | Use enzymes with low bias and good processivity. |
| Mismatch-Tolerant Aligner Software | Maps sequencing reads containing T-to-C mutations to the reference genome. | Key for downstream analysis (e.g., Bowtie, STAR). |
The study of RNA-binding proteins (RBPs) is crucial for understanding post-transcriptional gene regulation. Crosslinking and immunoprecipitation (CLIP) methods, including HITS-CLIP and PAR-CLIP, have been foundational. However, a persistent technical artifact known as truncation artifacts or "false truncations" arises from incomplete reverse transcription at the protein-RNA crosslink site, leading to cDNA fragments that map upstream of the actual crosslink. This obscures the precise identification of the protein-RNA interaction site. iCLIP (individual-nucleotide resolution CLIP) innovatively addresses this by introducing a cDNA circularization step, transforming a limitation into a precise mapping tool.
Core Mechanism: From Truncation to Circularization
In standard CLIP protocols, truncated cDNAs are discarded as waste. iCLIP repurposes them. The protocol is designed so that truncated cDNAs, which terminate at the crosslinked nucleotide, carry a unique adapter at their 3' end. After adapter ligation to the 5' end, the cDNA is circularized. During circularization, the 3' adapter becomes adjacent to the 5' adapter. PCR amplification across this novel junction creates a library where the position of the truncation (the crosslink site) is encoded within the sequence read as a mutation or a shift in the read start.
Quantitative Data Comparison: CLIP Methodologies
Table 1: Comparison of Key High-Resolution CLIP Variants
| Feature | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslinking Method | UV-C (254 nm) | UV-A (365 nm) + 4-thiouridine | UV-C (254 nm) |
| Resolution | ~30-60 nt (cluster) | ~20-30 nt (mutation site) | Single-nucleotide (cDNA start) |
| Key Signal | cDNA cluster boundaries | T-to-C transitions in reads | Truncation site via circularization |
| Handles Truncations? | No; treats as noise | Partially; mutations can mark site | Yes; leverages them for precision |
| Primary Artifact | Truncation artifacts | Photo-activation side effects | Circularization efficiency |
| Typical Read Yield | 10-50 million | 10-30 million | 5-20 million |
Table 2: Impact of iCLIP Circularization on Data Fidelity
| Metric | Without Circularization (Standard CLIP) | With iCLIP Circularization |
|---|---|---|
| Precise Crosslink Site ID | Ambiguous, broad peaks | Directly encoded in read |
| % of Mapped Reads Originating from Truncation Events | Lost or misassigned | ~50-80% (usable signal) |
| Background Noise from Truncations | High | Low (converted to signal) |
| Mapping Ambiguity at Binding Sites | High | Significantly Reduced |
Detailed iCLIP Protocol with cDNA Circularization
Part A: In Vivo Crosslinking, RNA Fragmentation, and Immunoprecipitation
- UV-C Crosslinking (254 nm): Irradiate cells or tissue to covalently link RBPs to their bound RNA.
- Cell Lysis & Partial RNase Digestion: Lyse cells and treat with low-concentration RNase I to produce protein-bound RNA fragments of ~50-70 nt.
- Immunoprecipitation: Use specific antibodies against the RBP of interest to purify RNP complexes. Perform stringent washes.
- 3' Adapter Ligation (Splinted): On-bead, ligate a pre-adenylated 3' DNA adapter to the RNA fragment's 3' end using T4 RNA Ligase 4. A DNA splint ensures specificity.
Part B: Reverse Transcription and Critical Circularization
- Reverse Transcription (RT): Prime RT with an oligonucleotide complementary to the 3' adapter. RT will terminate at the crosslinked nucleotide, producing a truncated cDNA. A second, non-truncated cDNA product may also form.
- Proteinase K Treatment & RNA Hydrolysis: Digest the protein to release cDNA:RNA hybrids. Hydrolyze the RNA with alkali.
- cDNA Purification: Isolate cDNAs via denaturing PAGE, selecting the size range corresponding to truncated and full-length products.
- 5' Adapter Ligation: Ligate a DNA adapter to the 5' end of the purified cDNA using T4 RNA Ligase 1.
- cDNA Circularization (Key Step): Use Circligase ssDNA Ligase to circularize the single-stranded, adapter-flanked cDNA.
- Reaction: Purified cDNA, Circligase buffer, ATP, betaine, Circligase enzyme. Incubate at 60°C for 1-2 hours.
- Principle: The enzyme joins the 3' and 5' adapters. For truncated cDNAs, this creates a circle where the junction between the original 3' and 5' ends now lies adjacent to the site of the crosslink/truncation.
Part C: PCR Amplification and Sequencing
- Linearization by PCR: Design PCR primers complementary to the adapter sequences now juxtaposed by circularization. Amplification linearizes the circle.
- Library Amplification & Sequencing: Perform limited-cycle PCR to generate the final sequencing library. Paired-end sequencing is standard. The read start corresponds precisely to the crosslink-induced truncation site.
Visualization of Workflows and Concepts
Diagram 1: Truncation Artifact vs iCLIP Solution (80 chars)
Diagram 2: iCLIP Molecular Steps to Encode Site (78 chars)
The Scientist's Toolkit: Key iCLIP Reagents
Table 3: Essential Research Reagent Solutions for iCLIP
| Reagent | Function & Role in Addressing Truncations |
|---|---|
| RNase I (Low Concentration) | Generates short RNA footprints bound by the RBP. Optimal fragmentation is critical for resolution. |
| Pre-adenylated 3' DNA Adapter | Substrate for T4 RnI4 ligation. Pre-adenylated prevents adapter self-ligation, ensuring single adapter addition. |
| T4 RNA Ligase 4 (RnI4) | Specifically ligates the pre-adenylated adapter to RNA 3' ends, crucial for initial library construction. |
| Circligase ssDNA Ligase | Core innovative enzyme. Circularizes single-stranded DNA, enabling the conversion of the truncation point into a sequenceable junction. |
| Betaine | Additive in the circularization reaction that enhances Circligase efficiency by reducing secondary structure in the cDNA. |
| Phusion High-Fidelity DNA Polymerase | Used for the final library PCR due to its high fidelity and processivity for amplifying circularized templates. |
| PAGE Gel Purification Reagents | Critical for size-selective purification of cDNAs after RT and before circularization, removing contaminants. |
| Proteinase K | Essential for digesting the crosslinked protein after RT, releasing the cDNA for subsequent steps. |
iCLIP's cDNA circularization strategy represents a paradigm shift in CLIP methodology. By ingeniously repurposing reverse transcription truncations—once a major source of noise—into the primary signal for single-nucleotide resolution, it provides a more accurate map of protein-RNA interactions. This protocol refinement, set within the broader thesis of evolving CLIP technologies (HITS-CLIP, PAR-CLIP), has become a cornerstone for rigorous RBP research, offering drug development professionals a clearer view of potential regulatory targets.
Key Commonalities and Philosophical Divergences Among the Three Major Protocols
Application Notes
The study of RNA-binding proteins (RBPs) is fundamental to understanding post-transcriptional gene regulation. The three major crosslinking and immunoprecipitation (CLIP) protocols—HITS-CLIP, PAR-CLIP, and iCLIP—are indispensable tools for mapping RBP-RNA interactions in vivo. These methods share a common conceptual framework but differ in their crosslinking chemistry and library preparation strategies, leading to distinct biases, resolutions, and applications. This document, framed within a thesis on CLIP-Seq advancements, delineates their core commonalities, philosophical divergences, and practical applications for researchers and drug development professionals targeting RBPs therapeutically.
Key Commonalities: All three protocols are designed to capture in vivo RBP-RNA interactions with nucleotide resolution. The shared workflow involves: (1) In vivo crosslinking of RBPs to their bound RNAs, (2) Cell lysis and partial RNA fragmentation, (3) Immunoprecipitation of the RBP-RNA complex under stringent conditions, (4) RNA linker ligation, (5) Protein removal and RNA isolation, and (6) Library preparation for high-throughput sequencing. This core pipeline ensures the captured RNA fragments are derived from direct, physiologically relevant protein interactions.
Philosophical Divergences: The primary divergence lies in the crosslinking strategy, which fundamentally shapes the experimental outcome and data interpretation.
- HITS-CLIP uses 254 nm UV-C light to induce covalent bonds primarily between the RBP and pyrimidine bases (U/C), creating a "standard" crosslink.
- PAR-CLIP incorporates 4-thiouridine (4SU) or 6-thioguanosine (6SG) nucleoside analogs into nascent RNA. Crosslinking with 365 nm UV-A light is more efficient and induces specific T-to-C (4SU) or G-to-A (6SG) mutations in the sequenced cDNA, providing a positive identification mark for crosslinked sites.
- iCLIP uses 254 nm UV-C light (like HITS-CLIP) but its philosophical innovation is in the library preparation. It captures truncated cDNAs that often stop at the crosslink site, allowing precise mapping to a single nucleotide.
The choice of protocol involves trade-offs between crosslinking efficiency, mutation signature for background reduction, resolution, and compatibility with the biological system (e.g., 4SU incorporation in primary cells).
Quantitative Data Comparison
Table 1: Core Characteristics of Major CLIP Protocols
| Feature | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslink Type | UV-C (254 nm) | UV-A (365 nm) | UV-C (254 nm) |
| Nucleoside Analog | None | 4-thiouridine (4SU) / 6-thioguanosine (6SG) | None |
| Key Mutational Signature | Deletions, crosslink-induced mutations (low frequency) | T-to-C (4SU) or G-to-A (6SG) transitions (high frequency) | Truncated cDNAs, deletions |
| Primary Resolution | ~30-60 nt (cluster-based) | ~20-30 nt (mutation-based) | ~1 nt (truncation-based) |
| Crosslinking Efficiency | Moderate | High (due to photoreactive analog) | Moderate |
| Background Signal | Higher | Lowest (mutation filter) | Low (truncation filter) |
| Primary Data Identifier | cDNA start site clusters | Mutation clusters | cDNA truncation sites |
| Typical Sequencing Depth | 10-30 million reads | 10-30 million reads | 15-50 million reads |
Table 2: Practical Considerations for Protocol Selection
| Consideration | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Best For | Robust, established RBPs; tissue samples | High precision mapping; cultured cells | Single-nucleotide resolution; studying reverse transcriptase arrest |
| Major Advantage | No metabolic labeling required; versatile. | High signal-to-noise; unambiguous sites. | Highest resolution; identifies modified nucleotides. |
| Major Limitation | Lower precision; higher background. | Requires metabolic labeling; cytotoxic potential of analogs. | Complex library prep; lower yield. |
| Compatibility with Tissue/In Vivo | Excellent | Poor to Moderate | Good |
Detailed Experimental Protocols
Protocol 1: HITS-CLIP (High-Throughput Sequencing Crosslinking Immunoprecipitation)
Principle: Utilize 254 nm UV light to crosslink RBPs to RNA in vivo, followed by rigorous purification and sequencing. Key Steps:
- In Vivo Crosslinking: Wash cells with PBS and irradiate with 254 nm UV light (e.g., 400 mJ/cm²) on ice.
- Cell Lysis: Lyse cells in stringent lysis buffer (e.g., 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- Partial RNase Digestion: Treat lysate with limited RNase I (e.g., 0.01 U/µg) to fragment RNA to ~50-100 nt.
- Immunoprecipitation: Incubate with antibody-coupled magnetic beads overnight at 4°C. Wash stringently with high-salt and detergent buffers.
- 3' Dephosphorylation & Linker Ligation: Dephosphorylate RNA ends with PNK (no ATP). Ligate a pre-adenylated 3' DNA linker.
- 5' Phosphorylation & Linker Ligation: Label 5' ends with PNK and [γ-³²P]ATP. Ligate a 5' RNA linker.
- SDS-PAGE & Transfer: Run complex on NuPAGE gel, transfer to nitrocellulose, and expose to film. Excise the region corresponding to the RBP-RNA complex.
- Proteinase K Digestion & RNA Extraction: Elute RNA from membrane slice and digest with Proteinase K. Phenol-chloroform extract and ethanol precipitate RNA.
- Reverse Transcription & PCR: Reverse transcribe with primer complementary to 3' linker. PCR amplify with indexed primers.
- Sequencing: Purify library and sequence on an Illumina platform.
Protocol 2: PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation)
Principle: Incorporate 4SU into RNA, crosslink with 365 nm UV light to induce T-to-C mutations, and use mutations to identify binding sites. Key Steps:
- 4SU Incorporation: Culture cells in medium supplemented with 100 µM 4-thiouridine (4SU) for 12-16 hours.
- In Vivo Crosslinking: Wash cells and irradiate with 365 nm UV light (e.g., 0.15 J/cm²) on ice.
- Cell Lysis & Immunoprecipitation: Lyse and perform IP as in HITS-CLIP (Steps 2-4).
- On-Bead RNase Digestion & Dephosphorylation: After IP washes, perform RNase I digestion on beads. Dephosphorylate with PNK.
- 3' Linker Ligation: Ligate pre-adenylated 3' linker directly on beads.
- Radiolabeling: Use PNK and [γ-³²P]ATP to label RNA on beads.
- SDS-PAGE, Transfer, & Elution: As in HITS-CLIP (Step 7). Excise region and elute RNA.
- Proteinase K Digestion & RNA Extraction: As in HITS-CLIP (Step 8).
- 5' Linker Ligation & Reverse Transcription: Ligate 5' RNA linker. Reverse transcribe with a primer containing Illumina adapter sequences.
- Circularization & PCR: Circularize cDNA with Circligase. Re-linearize and PCR amplify.
- Sequencing: Sequence on Illumina. Identify crosslinked sites by T-to-C mutations in aligned reads.
Protocol 3: iCLIP (Individual-Nucleotide Resolution CLIP)
Principle: Use 254 nm UV crosslinking, but capture cDNAs that truncate at the crosslink site during reverse transcription, enabling single-nucleotide mapping. Key Steps:
- Crosslinking & Lysis: Perform in vivo UV-C crosslinking (254 nm) and cell lysis as in HITS-CLIP.
- Partial RNase Digestion & Immunoprecipitation: As in HITS-CLIP.
- 3' Linker Ligation on Beads: After stringent washes, ligate a pre-adenylated 3' linker with a cleavable group (e.g., ribonucleotide) directly to the RNA on beads.
- Proximal Ligation of 5' Adapter: A key innovation. Reverse transcribe on-bead. The reverse transcriptase often stops at the crosslinked nucleotide. Ligate a single-stranded DNA adapter to the cDNA 5' end (not the RNA) using SplintR ligase. This "proximal ligation" links the adapter directly to the cDNA that terminated at the crosslink site.
- Elution & SDS-PAGE: Elute RBP-RNA-cDNA complex, run on SDS-PAGE, and transfer to nitrocellulose. Excise the region of interest.
- Proteinase K Digestion & RNA/cDNA Recovery: Digest with Proteinase K to release the RNA-cDNA hybrid.
- Circularizing PCR (ircPCR): Circularize the single-stranded cDNA using Circligase. Use primers complementary to the ligated adapters to PCR amplify the library.
- Sequencing: Sequence. The truncation site in the read (the 5' end of the cDNA insert) marks the crosslinked nucleotide.
Diagrams
Title: CLIP Protocol Comparison: Core Workflow and Divergences
Title: Conceptual Resolution of RBP Binding Sites by CLIP Method
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CLIP Experiments
| Item | Function | Key Considerations |
|---|---|---|
| UV Crosslinker (254 nm & 365 nm) | Induces covalent bond between RBP and RNA. | Calibrated energy output is critical for efficiency and cell viability. |
| 4-Thiouridine (4SU) | Photoactivatable nucleoside analog for PAR-CLIP. | Cytotoxicity at high doses; optimization of concentration and incorporation time required. |
| RNase I | Fragments RNA to manageable sizes for IP and sequencing. | Titration is crucial to avoid over-digestion and loss of signal. |
| Magnetic Protein A/G Beads | Solid support for antibody-mediated immunoprecipitation. | Pre-clearing with lysate reduces non-specific binding. |
| RBP-Specific Antibody | Captures the protein-RNA complex of interest. | Critical: Must be high-affinity, specific, and CLIP-validated. |
| Pre-adenylated 3' Linker | Ligates to RNA 3' end without ATP; prevents circularization. | Required for all protocols. Contains barcodes for multiplexing. |
| T4 Polynucleotide Kinase (PNK) | Dephosphorylates RNA 3' ends; phosphorylates 5' ends for ligation or radiolabeling. | Used in multiple steps; mutant versions available for specific functions. |
| [γ-³²P] ATP | Radiolabels RNA 5' ends for visualization by autoradiography. | Enables precise excision of the correct complex from the membrane. Safety protocols required. |
| Proteinase K | Digests the RBP to release the crosslinked RNA for downstream steps. | Essential for liberating RNA from the proteinaceous complex. |
| SplintR Ligase (for iCLIP) | Ligates single-stranded DNA adapter to cDNA 5' end during proximal ligation. | High efficiency is key for iCLIP library yield. |
| Circligase ssDNA Ligase | Circularizes single-stranded cDNA (iCLIP, PAR-CLIP). | Enables amplification of truncated or short cDNAs. |
| High-Fidelity DNA Polymerase | Amplifies final cDNA library for sequencing. | Minimizes PCR bias and errors in the final library. |
From Theory to Bench: Step-by-Step Protocols for HITS-CLIP, PAR-CLIP, and iCLIP
The success of any CLIP-seq variant (HITS-CLIP, PAR-CLIP, iCLIP) is fundamentally determined by decisions made prior to protocol execution. Within a thesis on RNA-binding protein (RBP) biology, this phase dictates the biological relevance and reproducibility of findings. The choice of cellular context and the rigor of experimental design directly influence the ability to map authentic, functional RBP-RNA interactions, which are critical for downstream applications in drug discovery and mechanistic biology.
Quantitative Comparison of Model Systems
The selection of cell line or tissue is a trade-off between physiological relevance, experimental tractability, and RBP expression. Key quantitative factors are summarized below.
Table 1: Quantitative & Qualitative Metrics for Model System Selection
| Metric | Immortalized Cell Lines (e.g., HEK293, HeLa) | Primary Cells | In Vivo / Tissue Samples |
|---|---|---|---|
| Physiological Relevance | Low-Medium (transformed, aberrant pathways) | High (normal karyotype, tissue-specific) | Highest (native niche, heterogeneity) |
| RBP Expression Endogeneity | Variable; may overexpress or lack specific RBPs | High | High |
| Required Cell Number | 5x10^6 - 2x10^7 per CLIP (easily scalable) | 1x10^7 - 5x10^7 (limited expansion) | 50-100 mg tissue (sample access limited) |
| Growth Rate / Availability | High (unlimited propagation) | Low (finite lifespan) | Requires animal models or biopsies |
| Genetic Manipulability | High (transfection, CRISPR) | Medium-Low (challenging) | Low (requires transgenic models) |
| Inter-Experiment Variability | Low (clonal, homogeneous) | Medium (donor variability) | High (biological complexity) |
| Cost & Throughput | Low / High | Medium / Medium | High / Low |
Foundational Experimental Design Protocols
A robust design is required to distinguish signal from noise in CLIP-seq data. These protocols must precede crosslinking.
Protocol 3.1: Design of Appropriate Controls
Objective: To control for non-specific RNA background and UV crosslinking artifacts. Key Controls:
- Untagged / Wild-Type Control: Use the parental cell line lacking the epitope-tagged RBP. Process identically to the experimental sample. Essential for identifying antibody non-specificity.
- UV Crosslinking Control (-UV): Process an identical sample without 254 nm UV irradiation. Critical for assessing the background of non-covalently bound RNA recovered during immunoprecipitation.
- RNase Titration Control: A pilot experiment to determine the optimal RNase I concentration that yields protected footprints (30-70 nt) after partial digestion, avoiding over- or under-digestion.
- Method: Aliquot cell lysate from a test crosslinking. Treat with a dilution series of RNase I (e.g., 0.1, 0.5, 1.0 U/μL) for 3 min at 37°C. Stop with Proteinase K, isolate RNA, and analyze on a Bioanalyzer (Pico Chip).
Protocol 3.2: Validation of RBP Expression and Localization
Objective: To confirm endogenous or tagged RBP expression and subcellular localization relevant to the research hypothesis. Method (Western Blot & Fractionation):
- Harvest cells or homogenize tissue in lysis buffer.
- For cytoplasmic/nuclear fractionation, use a hypotonic lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl) with 0.1% NP-40. Centrifuge at 3000xg for 5 min. Supernatant = cytoplasmic fraction. Pellet (nuclei) is washed and sonicated in RIPA buffer.
- Resolve 20-50 μg of protein by SDS-PAGE, transfer to PVDF membrane.
- Probe with validated antibody against the RBP (or tag). Use GAPDH (cytoplasm) and Lamin B1 (nucleus) as fractionation controls.
- Quantify expression level relative to control cell lines/tissues.
Protocol 3.3: Pilot Immunoprecipitation (IP) Efficiency Test
Objective: To quantify the efficiency of the antibody for IP under CLIP-stringent wash conditions prior to full-scale experiment. Method:
- UV-crosslink 1x10^6 cells (254 nm, 400 mJ/cm²).
- Lyse cells in 1 mL of stringent IP buffer (e.g., 50 mM HEPES pH 7.5, 300 mM NaCl, 1% NP-40, 0.5% Na-Deoxycholate, 0.1% SDS, Protease Inhibitors).
- Split lysate: 10% for "Input," 90% for "IP."
- Pre-clear lysate with protein A/G beads for 30 min.
- Incubate with 2-5 μg of specific antibody or isotype control for 2 hrs at 4°C.
- Add beads, incubate 1 hr, wash 3x with high-salt wash buffer (50 mM HEPES, 500 mM NaCl, 1% NP-40, 0.1% SDS).
- Elute beads and Input in 1X Laemmli buffer. Analyze by Western Blot.
- Calculate IP Efficiency: (SignalIP / SignalInput) * (VolumeInput / VolumeIP) * 100%. Target >5% for a robust CLIP-seq experiment.
Visualization of Decision Pathways and Workflows
Title: Decision Pathway for CLIP-seq Model System Selection
Title: Integrated Workflow from Experimental Design to CLIP-seq
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for Pre-Protocol Validation & CLIP-seq
| Reagent / Solution | Function & Critical Role | Example Product / Note |
|---|---|---|
| Anti-FLAG M2 / HA / MYC Antibody | High-affinity antibodies for immunoprecipitation of epitope-tagged RBPs. Critical for reducing background versus endogenous antibodies. | Sigma F1804, CST 3724 |
| RNase I (Commercial Grade) | For partial, non-specific digestion of unprotected RNA to leave protein-bound footprints. Lot-to-lot consistency is vital. | ThermoFisher EN0601 |
| 4-Thiouridine (4SU) / 6-Thioguanosine (6SG) | Photosensitive nucleoside analogs for PAR-CLIP. Incorporated into RNA, inducing T-to-C transitions upon 365nm crosslinking. | Merck T4509 / G10350 |
| UV Crosslinkers (254nm & 365nm) | Precise energy delivery for covalent crosslinking (HITS-CLIP/iCLIP: 254nm; PAR-CLIP: 365nm). Calibration is essential. | Spectrolinker XL-1500 |
| Stringent IP/Wash Buffers (with 0.1% SDS) | Maintains RNA-protein integrity while removing non-specific interactions. High-salt (500mM NaCl) buffers reduce background. | Prepared fresh with DEPC-H₂O. |
| Protein A/G Magnetic Beads | Solid-phase support for antibody-based IP. Magnetic separation improves wash efficiency and reduces RNA loss. | Pierce 88802 / 88803 |
| RNase Inhibitor (SUPERase•In) | Protects RNA from degradation during all non-digestion steps prior to library construction. | ThermoFisher AM2696 |
| T4 PNK (Phosphatase- minus Mutant) | For iCLIP cDNA truncation at crosslink sites. Critical for single-nucleotide resolution mapping. | NEB M0236S |
| High-Sensitivity RNA/DNA Analysis Kits | For accurate quantification and size distribution analysis of input RNA and final libraries (Bioanalyzer/TapeStation). | Agilent 5067-1513 |
Metabolic labeling of nascent RNA with 4-thiouridine (4SU) is the defining step of the Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) method. Within the broader thesis of CLIP-Seq methodologies (HITS-CLIP, PAR-CLIP, iCLIP) for RNA-binding protein (RBP) research, this step introduces a specific, high-resolution T-to-C transition mutation signature in sequencing libraries, allowing for precise identification of crosslink sites. This application note details the optimization and critical controls for the 4SU labeling step, which is pivotal for successful PAR-CLIP experiments in basic research and drug discovery targeting RBPs.
Optimization Parameters for 4SU Labeling
Successful incorporation of 4SU requires balancing labeling efficiency with cellular toxicity. The key parameters for optimization are summarized below.
Table 1: Optimization Variables for Metabolic 4SU Labeling
| Parameter | Typical Range | Optimization Goal | Impact on Experiment |
|---|---|---|---|
| 4SU Concentration | 100 µM – 500 µM | Maximize incorporation while minimizing cytotoxicity. | Higher conc. increases crosslink efficiency but can perturb cell physiology. |
| Labeling Duration | 1 hour – 16 hours | Sufficient for RBP-bound transcript turnover. | Shorter times reduce toxicity; longer times ensure labeling of less abundant targets. |
| Cell Type / Line | Variable | Determine tolerance to 4SU and nucleoside transporters. | Primary cells are often more sensitive than immortalized lines. |
| Serum Concentration | 2% – 10% during labeling | Reduce serum competition for nucleoside uptake. | Lower serum (e.g., 2%) can enhance 4SU uptake but may stress cells. |
| Control: DMSO Vehicle | Equivalent volume | Account for solvent effects. | Essential negative control for gene expression changes. |
Table 2: Troubleshooting 4SU Labeling
| Problem | Potential Cause | Solution |
|---|---|---|
| Low T-to-C mutation rate | Insufficient 4SU incorporation; inefficient 365 nm crosslinking. | Increase 4SU concentration/duration; verify UV lamp energy. |
| High Cell Death | 4SU cytotoxicity; overly stringent serum reduction. | Titrate 4SU; use shorter pulse; maintain higher serum (5-10%). |
| High Background in Libraries | Non-specific RNA degradation or carryover. | Include a no-UV control; optimize RNase T1 concentration; stringent washing. |
| No RNA Recovery | Excessive cytotoxicity; RBP not binding 4SU-labeled RNA. | Verify cell viability post-labeling; consider alternative CLIP method (e.g., iCLIP). |
Detailed Protocol: Metabolic Labeling with 4SU
Materials
- Research Reagent Solutions:
- 4-Thiouridine (4SU) Stock Solution: 500 mM in DMSO. Store at -20°C protected from light. Function: Photoactivatable ribonucleoside for metabolic RNA labeling.
- Dimethyl Sulfoxide (DMSO): Cell culture grade. Function: Vehicle control for 4SU stock.
- Pre-warmed, Serum-free or Low-Serum Medium: Appropriate for cell line (e.g., 2% FBS). Function: Reduces competition for nucleoside transporters to enhance 4SU uptake.
- Phosphate-Buffered Saline (PBS), pre-warmed.
- Total RNA Extraction Reagent (e.g., TRIzol). Function: For assessing 4SU incorporation efficiency.
- 365 nm UV Crosslinker (e.g., Spectrolinker). Function: Activates 4SU to crosslink to bound RBPs.
Method
- Cell Preparation: Culture adherent or suspension cells to ~70-80% confluency.
- 4SU Administration:
- Prepare working medium: Dilute 4SU stock into pre-warmed, low-serum medium to the desired final concentration (e.g., 100 µM). For vehicle control, add equivalent volume of DMSO to separate medium.
- Aspirate growth medium from cells and gently wash once with pre-warmed PBS.
- Add the 4SU-containing or control medium to the cells.
- Incubate cells for the optimized labeling period (e.g., 4-16 hours) under standard growth conditions (37°C, 5% CO₂).
- Post-Labeling Wash & Crosslinking:
- Aspirate the 4SU medium. Wash cells twice with generous volumes of pre-warmed PBS.
- For adherent cells: Aspirate PBS, add a thin layer of PBS, and crosslink on ice using 365 nm UV light at 0.15 J/cm² (e.g., 150 mJ/cm² at 254 nm setting equivalence, or specific 365 nm setting).
- For suspension cells: Pellet, resuspend in PBS, crosslink in a Petri dish, then pellet again.
- No-UV Control: Process an equivalent sample identically but shield from 365 nm light. This is critical for identifying background.
- Cell Collection: After crosslinking, scrape or pellet cells. Flash-freeze cell pellets in liquid nitrogen and store at -80°C until lysis for PAR-CLIP.
Essential Controls for 4SU Labeling
- DMSO Vehicle Control: Label cells with DMSO alone to control for changes in gene expression or RBP activity due to the solvent.
- No-UV Control (-UV): Process 4SU-labeled cells identically but omit 365 nm crosslinking. This controls for non-covalent RNA-protein interactions and background in immunoprecipitation.
- Incorporation Efficiency Check: Isolate total RNA from a small aliquot of labeled cells. Measure the absorbance ratio A330/A260. An increase in A330 indicates 4SU incorporation.
Visual Workflow and Pathway
Diagram 1: PAR-CLIP 4SU Labeling Workflow & Molecular Outcome (85 chars)
Diagram 2: Research Reagent Toolkit for PAR-CLIP 4SU Labeling (65 chars)
Application Notes
In the study of RNA-binding proteins (RBPs) through CLIP-Seq variants (HITS-CLIP, PAR-CLIP, iCLIP), the initial steps of in vivo crosslinking, cell lysis, and RNase treatment form a critical, universal core. These steps determine the specificity and resolution of the final dataset by covalently capturing transient RNA-protein interactions, efficiently recovering complexes, and generating RNA footprints of optimal size. This protocol details a standardized and optimized approach for this universal core, emphasizing rigorous empirical RNase titration, which is paramount for balancing crosslink-site resolution against library complexity.
Key Quantitative Parameters for Core Steps
Table 1: Standardized Parameters for Universal Core Steps
| Step | Key Parameter | Typical Range | Optimization Notes |
|---|---|---|---|
| In Vivo UV Crosslinking | UV-C Energy (254 nm) | 150-400 mJ/cm² | 400 mJ/cm² common for standard CLIP; lower energy may reduce background. |
| Cell Type | Cultured cells, tissue | Tissue requires homogenization post-crosslink. | |
| Cell Lysis & Clarification | Lysis Buffer Volume | 1 mL per 10⁷ cells | Ensure complete disruption. |
| Protease Inhibitors | 1x cocktail | Essential to prevent RBP degradation. | |
| RNase Inhibitors | 0.5-1 U/μL | Critical post-lysis until RNase step. | |
| Clarification (Centrifugation) | 16,000-20,000 x g, 15 min, 4°C | Removes nuclei, debris. | |
| Rigorous RNase Titration | RNase I Concentration | 0.001 - 0.1 U/μL | Must be determined empirically. See Table 2. |
| Digestion Temperature & Time | 37°C, 3-15 min | Constant for titration series. | |
| Post-digestion RNA Fragment Size | 50-100 nt (post-proteinase K) | Target range for library construction. |
Table 2: Empirical RNase Titration Scheme & Expected Outcomes
| RNase I Dilution (U/μL) | Digestion Time (min) | Expected RNA Fragment Size (nt) | Goal of Condition |
|---|---|---|---|
| 0.001 | 5, 10, 15 | >150 | Identify under-digestion point (low yield). |
| 0.01 | 5, 10, 15 | 70-120 | Target optimal range. |
| 0.05 | 5, 10, 15 | 40-70 | Identify over-digestion point (high background). |
| 0.1 | 5 | <50 | Control for over-digestion. |
Detailed Protocols
Protocol 1: In Vivo UV Crosslinking (254 nm) for Adherent Cells
Materials: PBS (ice-cold), UV crosslinker (254 nm), cell scraper, microcentrifuge tubes.
- Preparation: Aspirate culture medium from adherent cells (one 15-cm dish per condition). Wash cells twice with 10 mL of ice-cold PBS.
- Crosslinking: Aspirate PBS completely. Place dish, lid removed, in a UV crosslinker pre-calibrated to 400 mJ/cm² (or optimized energy). Perform crosslinking.
- Harvest: Immediately add 1 mL of ice-cold PBS to the dish. Scrape cells and transfer the suspension to a pre-chilled 1.5 mL microcentrifuge tube.
- Pellet: Centrifuge at 700 x g for 5 min at 4°C. Aspirate supernatant. Cell pellet can be flash-frozen in liquid N₂ or processed immediately for lysis.
Protocol 2: Denaturing Cell Lysis and Clarification
Lysis Buffer (make fresh): 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40 or Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate, 1x protease inhibitor (EDTA-free), 1 U/μL RNase inhibitor, 1 mM DTT.
- Resuspend cell pellet (~10⁷ cells) in 1 mL of ice-cold lysis buffer by vigorous pipetting.
- Incubate on ice for 15 min with occasional vortexing.
- Sonicate the lysate briefly (3 x 5 sec pulses, 30% amplitude) to shear genomic DNA and reduce viscosity. Keep samples on ice between pulses.
- Clarify the lysate by centrifugation at 20,000 x g for 15 min at 4°C.
- Carefully transfer the supernatant (cleared lysate) to a new pre-chilled tube. Measure protein concentration. Proceed immediately to RNase treatment or flash-freeze in aliquots.
Protocol 3: Rigorous Empirical RNase I Titration and Partial Digestion
Materials: Cleared cell lysate, RNase I (dilution series prepared in nuclease-free water), 200 U/μL SUPERase•In RNase Inhibitor, Proteinase K buffer.
- Aliquot 100 µL of cleared lysate (containing ~1-2 mg total protein) into four separate tubes labeled A-D.
- Prepare a 10x RNase I master mix series from a stock (e.g., 1 U/µL) in nuclease-free water: Tube A (0.01 U/µL), Tube B (0.05 U/µL), Tube C (0.1 U/µL), Tube D (0 U/µL - No RNase control).
- Add 11 µL of the appropriate 10x RNase I dilution to each corresponding lysate aliquot. Mix quickly by pipetting.
- Incubate all tubes at 37°C for exactly 10 minutes in a thermal mixer.
- Immediately stop the digestion by adding 11 µL of SUPERase•In RNase Inhibitor (200 U/µL) to each tube. Mix and place on ice.
- For analysis, remove a 20 µL sample from each condition and the no-RNase control. Add Proteinase K, extract RNA, and analyze fragment size distribution on a Bioanalyzer (Agilent) or Tapestation (Agilent).
- Select the optimal condition that yields the majority of RNA fragments in the 50-100 nt range post-proteinase K treatment for use in the subsequent immunoprecipitation steps of your chosen CLIP protocol.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Universal Core CLIP Steps
| Reagent / Solution | Function & Importance | Key Considerations |
|---|---|---|
| UV Crosslinker (254 nm) | Covalently links RBPs to bound RNA in vivo. | Calibrated energy output is critical for reproducibility. |
| Denaturing Lysis Buffer | Extracts crosslinked RNP complexes while inhibiting endogenous RNases and proteases. | SDS and deoxycholate denature proteins; NP-40 aids solubilization. |
| RNase I | Partially digests RNA not protected by the crosslinked RBP to generate footprints. | Enzyme activity lot-to-lot variability necessitates empirical titration. |
| SUPERase•In RNase Inhibitor | Irreversibly inactivates RNase I after digestion to halt reaction precisely. | More effective than RNasin for stopping RNase I. |
| Proteinase K | Digests proteins after immunoprecipitation to release crosslinked RNA fragments. | Essential for reversing crosslinks and RNA recovery. |
| Agilent Bioanalyzer/Tapestation | Provides high-sensitivity electrophoregrams of RNA fragment size distribution. | Critical tool for evaluating RNase titration results. |
Diagrams
Title: Universal Core Workflow for CLIP-Seq
Title: Empirical RNase Titration Process
Within the framework of CLIP-Seq methodologies (HITS-CLIP, PAR-CLIP, iCLIP), successful identification of RNA-protein interactions hinges on the specificity of the immunoprecipitation (IP) step. This IP crucible—where antibody, bead, and wash stringency converge—determines the signal-to-noise ratio in subsequent sequencing. Imperfections here propagate, obscuring true binding sites. This application note details protocols and considerations to optimize this core step for RBP research and drug discovery.
Antibody Validation: The Primary Specificity Check
The choice of antibody is the most critical variable. For CLIP, antibodies must be validated for use in IP under denaturing conditions.
Key Validation Criteria:
- Application-Specific: Validation for IP or chromatin IP (ChIP) is required; western blot validation alone is insufficient.
- Knockout/Knockdown Control: The gold standard. IP should show significant loss of signal in matched cell or tissue lysates from RBP knockout models.
- Cross-Reactivity: Assessment via mass spectrometry or western blot to identify off-target protein binding.
Table 1: Antibody Validation Strategies & Metrics
| Validation Method | Protocol Summary | Key Quantitative Metric | Acceptance Threshold |
|---|---|---|---|
| Genetic Knockout/Knockdown | Perform parallel IP from WT and KO lysates. Detect co-precipitated RNA (radiolabel or qPCR) or protein (western). | Signal Enrichment (KO vs WT) | >90% signal reduction in KO |
| Cross-Reactivity Profiling (MS) | Submit IP eluates for label-free quantitative mass spectrometry. | Spectral Counts for Target vs. Top Non-Target | ≥10-fold enrichment of target |
| Tagged Protein Rescue | IP against tag on exogenous, expressed RBP in KO background. | Comparison of IP efficiency between tag and native antibody. | Comparable or superior recovery |
Protocol: Antibody Validation using Knockout Lysates
- Prepare Lysates: Harvest wild-type (WT) and RBP knockout (KO) cells (e.g., CRISPR-generated) in identical IP lysis buffer (e.g., 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- Pre-clear: Incubate 500 µg of each lysate with 20 µL of bare beads for 30 min at 4°C. Pellet beads, collect supernatant.
- Immunoprecipitation: Split each pre-cleared lysate (250 µg each). To one, add 2 µg of target antibody. To the other, add 2 µg of species-matched IgG. Incubate 2 hours at 4°C.
- Capture: Add 25 µL of pre-washed Protein A/G beads. Incubate 1 hour.
- Wash & Elute: Wash beads 3x with 1 mL lysis buffer. Elute protein with 30 µL 2X Laemmli buffer at 95°C for 10 min.
- Analysis: Run eluates by SDS-PAGE. Perform western blot for the target RBP. Signal should be present in the WT antibody lane and absent in all KO lanes and IgG controls.
Bead Choices: The Capture Matrix
Bead composition impacts background binding and ligand accessibility.
Table 2: Bead Type Comparison for CLIP Protocols
| Bead Type | Surface Chemistry | Binding Capacity | Pros for CLIP | Cons for CLIP |
|---|---|---|---|---|
| Magnetic Protein A/G | Recombinant Protein A and/or G covalently coupled. | ~10-50 µg IgG/mL beads | Rapid separation, low non-specific RNA binding. | Potential for antibody leaching under harsh washes. |
| Agarose Protein A/G | Protein A/G cross-linked to agarose. | ~20-40 µg IgG/mL beads | High chemical/thermal stability, robust for stringent washes. | Slower centrifugation steps, potential for trapped RNA. |
| Magnetic Tosylactivated | Activated surface for covalent antibody coupling. | Varies by coupling. | Antibody not co-eluted, allowing cleaner RNA recovery; ideal for quantitative applications. | Additional coupling steps required; antibody cannot be reused. |
Stringency Washes: Balancing Specificity and Yield
Stringent washing removes non-specifically bound RNA while preserving true RBP-RNA complexes.
Core Wash Buffers:
- High-Salt Wash: 50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS. Disrupts ionic interactions.
- Denaturing Wash: 50 mM Tris-HCl pH 7.4, 500 mM LiCl, 1 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS. Disrupts hydrophobic & protein-protein interactions.
- Urea Wash (iCLIP): 50 mM Tris-HCl pH 7.4, 500 mM LiCl, 1 mM EDTA, 0.5% Urea. Mild denaturant to reduce background.
Standard CLIP Stringency Wash Protocol:
- After antibody-bead capture, pellet beads and aspirate supernatant.
- Wash 1: 1 mL of standard IP lysis buffer. Invert tube 10x. Pellet, aspirate.
- Wash 2: 1 mL of High-Salt Wash buffer. Invert for 2 minutes at room temperature. Pellet, aspirate.
- Wash 3: 1 mL of Denaturing Wash buffer. Invert for 2 minutes at room temperature. Pellet, aspirate.
- Wash 4: 1 mL of High-Salt Wash buffer again. Pellet, aspirate.
- Final Wash: 1 mL of 1X T4 PNK buffer (or equivalent). Pellet, remove all supernatant. Proceed to on-bead RNA processing (e.g., phosphorylation, linker ligation).
Visualizations
CLIP IP Core Workflow
Antibody Quality Dictates IP Outcome
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for the CLIP IP Crucible
| Reagent/Material | Function & Role in IP | Example Product/Catalog |
|---|---|---|
| Validated Antibody | Specifically captures target RBP and its crosslinked RNA. | Cell Signaling Technology, Abcam (KO-validated) |
| Magnetic Protein A/G Beads | Solid-phase matrix for efficient antibody-antigen capture and washing. | Pierce Magnetic Protein A/G, Dynabeads |
| RNase Inhibitor | Preserves RNA integrity during lysis and IP steps. | SUPERase•In, RNasin |
| Protease Inhibitor Cocktail | Maintains protein integrity and antibody epitopes. | EDTA-free tablets (e.g., Roche) |
| Mild Crosslinker | Stabilizes transient RBP-RNA interactions in vivo. | UV-C (254nm) for HITS/iCLIP; 4-Thiouridine + 365nm UVA for PAR-CLIP |
| High-Salt & Denaturing Wash Buffers | Removes non-specifically bound RNA and proteins post-IP. | Custom formulations per protocol. |
| RNase I (for some protocols) | Trims exposed RNA not protected by the bound RBP to leave footprint. | Ambion RNase I |
| T4 PNK (Polynucleotide Kinase) | Critical for RNA end repair and radiolabeling in CLIP workflows. | NEB T4 PNK |
| Covalent Coupling Kit (Optional) | For coupling antibodies to tosylactivated beads. | Abcam Antibody Coupling Kit |
The precise preparation of sequencing libraries is the critical determinant of success in UV crosslinking and immunoprecipitation (CLIP) methodologies, including HITS-CLIP, PAR-CLIP, and iCLIP. These protocols, fundamental for in vivo RNA-binding protein (RBP) research and drug target discovery, rely on the efficient conversion of a single, crosslinked RNA-protein adduct into a sequenceable DNA molecule. The nuances of adapter ligation, reverse transcription, and PCR amplification directly impact the fidelity, complexity, and bias of the final dataset, influencing downstream biological conclusions.
Key Steps and Quantitative Comparisons
The library preparation workflow for CLIP-seq variants shares a common skeleton but exhibits crucial, protocol-specific differences, primarily in adapter design and handling of cDNA truncation events. The quantitative parameters for core enzymatic steps are summarized below.
Table 1: Comparative Parameters for Library Preparation Steps in Major CLIP Protocols
| Step / Parameter | HITS-CLIP / CLIP-seq | PAR-CLIP | iCLIP | Functional Rationale |
|---|---|---|---|---|
| RNA 3' Adapter Ligation | Pre-calibration of T4 RNA Ligase 1 (truncated, K227Q) activity; High [ATP] (1 mM) | Pre-calibration of T4 RNA Ligase 1 (truncated, K227Q) activity | Ligation with T4 RNA Ligase 2 (truncated, KQ) | Minimizes circularization of RNA; KQ mutants lack adenylation activity, reducing adapter multimer formation. |
| Reverse Transcription | Standard primer extension with Superscript III/IV | Standard primer extension | Template-switching using TGIRT or SuperScript II | iCLIP uses template-switching to add a universal sequence at cDNA 5' end, bypassing inefficient RNA 5' adapter ligation. |
| cDNA Purification & Size Selection | Denaturing PAGE (6-10% Urea gel); excision of ~70-100 nt region above linker-adapter | Denaturing PAGE; excision based on expected shift from T-to-C transitions | Denaturing PAGE; isolation of full-length and truncated cDNAs | Removes unextended primers, linker-linker ligation products, and selects for cDNA derived from crosslinked RNA fragments. |
| cDNA 3' Adapter Ligation | Circligase ssDNA Ligase | Circligase ssDNA Ligase | Not Required | iCLIP adapter is introduced during RT via template-switching. Circularization (HITS/PAR) protects cDNA ends. |
| PCR Amplification | 12-18 cycles with Phusion/UDPI; dual-indexed primers | 12-18 cycles; primers compatible with T-to-C coding | 10-15 cycles; primers for template-switch sequence | Limited cycle number prevents over-amplification bias; indexing enables multiplexing. PAR-CLIP primers must avoid reverse complementarity to mutated sites. |
Detailed Experimental Protocols
Protocol 3.1: Optimized 3' RNA Adapter Ligation for HITS-CLIP/PAR-CLIP
- Input: RNA recovered from stringent IP washes (on beads).
- Reagents: T4 RNA Ligase 1 (truncated K227Q, NEB), 10X Ligase Buffer, 25% PEG-8000, High-purity 3' DNA adapter (with 5' App & 3' blocking group), RNasin.
- Procedure:
- Assemble on ice: 5 µL RNA beads, 1 µL 10X Ligase Buffer, 1 µL 25% PEG-8000, 1 µL 3' DNA Adapter (10 µM), 1 µL RNasin (40 U/µL), 1 µL T4 RnL1 (trunc KQ) (10 U/µL). Total 10 µL.
- Incubate: 22°C for 2 hours in a thermomixer with gentle agitation (300 rpm).
- Wash beads 3x with stringent wash buffer. Proceed to on-bead reverse transcription.
Protocol 3.2: Reverse Transcription with Template-Switching for iCLIP
- Input: RNA beads post 3' adapter ligation.
- Reagents: TGIRT-III enzyme (InGex) or SuperScript II, corresponding buffer, dNTPs, Template-Switch Oligo (TSO), RNase Inhibitor.
- Procedure (TGIRT):
- Prepare RT mix: 1 µL 10X TGIRT Buffer, 1 µL DTT (100 mM), 0.5 µL dNTPs (10 mM each), 0.5 µL RNase Inhibitor, 1 µL RT primer (10 µM), 1 µL TSO (10 µM), 3 µL Nuclease-free water, 2 µL TGIRT-III enzyme (200 U). Total 10 µL.
- Add mix to 5 µL washed RNA beads. Total 15 µL.
- Incubate: 60°C for 30 min (TGIRT) or 42°C for 50 min (SSII).
- Immediately place on ice. Treat with 1 µL RNase I (10 U) for 5 min at 37°C to digest non-crosslinked RNA.
- Wash beads 2x with high-salt buffer.
Protocol 3.3: cDNA Circularization for HITS-CLIP/PAR-CLIP
- Input: Purified cDNA eluted from gel slice.
- Reagents: Circligase II ssDNA Ligase (Lucigen), 10X Circligase Buffer, Betaine, MnCl₂.
- Procedure:
- Assemble reaction: 5 µL purified cDNA, 1 µL 10X Circligase Buffer, 0.5 µL 50 mM MnCl₂, 2.5 µL 5M Betaine, 0.5 µL Circligase II (100 U/µL). Total 9.5 µL.
- Incubate: 60°C for 1-2 hours.
- Heat-inactivate: 80°C for 10 min.
- Use 2-5 µL directly for PCR.
Protocol 3.4: Limited-Cycle PCR Amplification
- Input: Circularized cDNA (HITS/PAR) or linear cDNA (iCLIP).
- Reagents: Phusion High-Fidelity DNA Polymerase (NEB), 5X HF Buffer, dual-indexed P5/P7 primers.
- Procedure:
- Prepare master mix on ice: 5 µL 5X HF Buffer, 0.5 µL dNTPs (10 mM), 0.5 µL P5 primer (10 µM), 0.5 µL P7 primer (10 µM), 0.25 µL Phusion polymerase, 13.25 µL Nuclease-free water. Total 20 µL per reaction.
- Add 5 µL of cDNA template.
- Run PCR: 98°C 30s; [98°C 10s, 60°C 30s, 72°C 20s] x N cycles; 72°C 5 min. Hold at 4°C. (N=12-18, determined by qPCR side-reaction).
- Purify PCR product with SPRI beads (1.8x ratio). Quantify by Bioanalyzer/Qubit.
Visualizations
Title: CLIP-seq Library Preparation Core Workflow Comparison
Title: iCLIP Reverse Transcription Mechanism
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CLIP-seq Library Construction
| Reagent / Kit Component | Vendor Examples | Function in Protocol |
|---|---|---|
| T4 RNA Ligase 1, truncated K227Q | NEB (M0437) | Catalyzes 3' adapter ligation with reduced RNA circularization and adapter dimerization. |
| App-modified 3' DNA Adapter | IDT, Sigma | Contains 5' adenylation (App) and 3' blocking group (e.g., amine) to ensure single, directional ligation. |
| TGIRT-III Reverse Transcriptase | InGex | Group II intron-derived RT with high processivity and template-switching efficiency, crucial for iCLIP. |
| Circligase II ssDNA Ligase | Lucigen (CL9021K) | Efficiently circularizes single-stranded cDNA, protecting molecule ends and enabling PCR amplification. |
| Phusion High-Fidelity DNA Pol | Thermo Fisher (F530) | High-fidelity polymerase for limited-cycle PCR, minimizing amplification errors in final library. |
| Urea-PAGE Gel System (6-10%) | Invitrogen, C.B.S. | Critical size-selection step to isolate cDNA of correct length and remove enzymatic reaction contaminants. |
| RNase Inhibitor (Murine) | Promega (N2615) | Protects RNA fragments on beads from degradation during enzymatic steps prior to reverse transcription. |
| SPRIselect Beads | Beckman Coulter (B23318) | For consistent size-selection and clean-up of PCR-amplified libraries prior to sequencing. |
Within the study of RNA-binding proteins (RBPs) using UV-crosslinking and immunoprecipitation (CLIP) methods (HITS-CLIP, PAR-CLIP, iCLIP), sequencing parameter selection is critical for robust, reproducible, and biologically meaningful data. This application note details considerations for sequencing depth, read length, and replicate strategy, grounded in current best practices for CLIP-seq experiments.
Key Sequencing Parameters
Sequencing Depth
Required depth varies by CLIP variant and biological question. Insufficient depth misses low-affinity binding sites, while excessive depth yields diminishing returns.
Table 1: Recommended Sequencing Depths for CLIP Methods
| Method | Typical Minimum Depth (M reads) | Recommended Depth for Saturation (M reads) | Primary Determinants |
|---|---|---|---|
| HITS-CLIP | 10-15 | 20-30 | RBP abundance, binding site distribution |
| PAR-CLIP | 8-12 | 15-25 | Mutation rate, crosslinking efficiency |
| iCLIP | 15-20 | 25-40 | cDNA truncation efficiency, library complexity |
Read Length
Read length must accommodate the fragmented RNA footprints and necessary adapters.
Table 2: Read Length Considerations
| Consideration | Single-End (SE) | Paired-End (PE) |
|---|---|---|
| Typical Length | 50-75 bp | 50-75 bp (Read 1) + 25-50 bp (Read 2) |
| Main Advantage | Cost-effective, sufficient for mapping | Identifies PCR duplicates more accurately, can span longer fragments |
| Recommended for | Standard, high-abundance RBP studies | Complex or repetitive genomes, duplicate removal critical |
Replicate Strategy
Biological replicates are non-negotiable for statistical rigor and reproducibility. Technical replicates (library prep from same sample) are less critical than biological replicates (independent samples).
Table 3: Replicate Strategy Guidelines
| Replicate Type | Minimum Number | Purpose | Key Analysis Use |
|---|---|---|---|
| Biological | 2-3 | Capture biological variability, assess reproducibility | Identify high-confidence binding sites via concordance (e.g., IDR). |
| Experimental Control | 1-2 per condition | Control for non-specific background | Input, IgG, or RNase-treated controls for peak calling. |
Detailed Experimental Protocols
Protocol 1: Library QC and Sequencing Depth Pilot
Objective: Determine the saturation point for a given RBP experiment. Materials: Prepared CLIP library, Bioanalyzer/TapeStation, qPCR quantitation kit, sequencer. Procedure:
- Quantify Library: Precisely quantify final library using fluorometric assay (e.g., Qubit) and qPCR for amplifiable molecules.
- Profile Size: Analyze library on High Sensitivity DNA chip to confirm expected fragment size distribution (typically 70-200 bp).
- Sequencing Run: Sequence a pilot fraction (e.g., 10-15% of a lane) to ~5-8 million reads.
- Saturation Analysis:
a. Map reads to the reference genome.
b. Use tools like
UMI-toolsorCLIPperto deduplicate using Unique Molecular Identifiers (UMIs). c. Subsample the mapped reads at increasing intervals (10%, 20%...100%). d. Plot the number of unique, non-redundant binding sites identified at each depth. e. The point where the curve plateaus indicates saturation depth.
Protocol 2: Biological Replicate Preparation for iCLIP
Objective: Generate two independent iCLIP libraries from distinct biological samples. Materials: Cultured cells or tissue, iCLIP lysis buffer, RNase I, PNK, specific antibody, Proteinase K, reverse transcription primers, circularization ligase. Procedure: Note: Use identical reagents and lot numbers for replicates.
- Crosslinking & Harvest: UV-crosslink cells (254 nm, 150-400 mJ/cm²). Harvest cells separately for each replicate.
- Cell Lysis and RNase Digestion: Lyse cells in stringent buffer. Treat lysate with optimal RNase I concentration (determined empirically) to generate ~50-100 nt footprints.
- Immunoprecipitation (IP): Incubate lysate with antibody-coated beads. Wash stringently.
- 3' Dephosphorylation & Ligation: Dephosphorylate RNA with PNK (no ATP). Ligate pre-adenylated 3' adapter.
- 5' Phosphorylation & Ligation: Phosphorylate 5' end with PNK (+ATP). Ligate 5' RNA adapter.
- RNA Isolation & Reverse Transcription: Purify RNA, reverse transcribe with primers containing Illumina sequences and sample barcodes.
- cDNA Circularization & PCR: Circularize cDNA. Amplify with PCR using indexing primers for 10-15 cycles.
- Library QC: Purify and quantify libraries as in Protocol 1. Pool replicates at equimolar ratios for sequencing.
Protocol 3: Background Signal Assessment with Control Replicates
Objective: Generate an IgG control library for peak-calling background subtraction. Materials: Non-specific species-matched IgG, all reagents from Protocol 2. Procedure:
- Perform the full iCLIP protocol (Protocol 2) in parallel, substituting the specific RBP antibody with an equal amount of control IgG.
- Sequence the control library to a depth equivalent to 50-100% of the experimental sample depth.
- Use peak-calling software (e.g.,
CLIPper,Piranha) that incorporates the control track to filter out peaks present in both IP and control.
Diagrams
Diagram 1: CLIP-seq Experimental & Sequencing Workflow
Diagram 2: Determining Sequencing Saturation
Diagram 3: Replicate Strategy Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Robust CLIP-seq Studies
| Reagent Category | Specific Example(s) | Function & Importance |
|---|---|---|
| Crosslinker | UV-C light (254 nm) | Covalently binds RBP to RNA at "zero-length". Critical for snapshot of in vivo interactions. |
| RNase | RNase I (Ambion) | Generates specific RNA fragment sizes. Lot-to-lot consistency is crucial for reproducibility. |
| Specific Antibody | Validated RBP antibody (e.g., from Sigma, Abcam) | Enriches for RBP-RNA complexes. Specificity is the single most important factor. |
| Control Antibody | Species-matched IgG | Distinguishes specific signal from background in IP. Essential for peak calling. |
| Adapter Oligos | Pre-adenylated 3' adapter (IDT), 5' RNA adapter | Enable sequencing library construction. Use HPLC-purified oligos. |
| UMI Adapters | Adapters containing random molecular barcodes (e.g., NNNNNN) | Allows bioinformatic removal of PCR duplicates, improving accuracy of depth calculations. |
| Reverse Transcriptase | SuperScript IV (Invitrogen) | High processivity and fidelity for reading through crosslinked sites, especially in iCLIP. |
| High-Fidelity Polymerase | KAPA HiFi HotStart ReadyMix | Low-error PCR amplification of final library to maintain sequence diversity. |
| Library Quant Kit | KAPA Library Quantification Kit (Roche) | Accurate qPCR-based quantification of amplifiable fragments for precise pooling. |
Within the broader thesis on experimental evolution of RNA-binding protein (RBP) mapping—encompassing CLIP-Seq, HITS-CLIP, PAR-CLIP, and iCLIP protocols—enhanced CLIP (eCLIP) represents a pivotal refinement. This protocol addresses critical limitations of predecessor methods, primarily high background noise and low signal-to-noise ratio, enabling more robust and reproducible identification of RBP-RNA interactions. The subsequent drive towards single-nucleotide resolution mapping marks the frontier in dissecting the precise mechanistic roles of RBPs in gene regulation, splicing, and disease pathogenesis, with direct implications for therapeutic target identification in drug development.
Core Refinements of the eCLIP Protocol
The eCLIP protocol introduced key modifications to the traditional iCLIP framework, significantly enhancing specificity and yield.
Table 1: Key Modifications in eCLIP vs. Standard iCLIP
| Protocol Step | Standard iCLIP | eCLIP Refinement | Primary Impact |
|---|---|---|---|
| UV Crosslinking | 254 nm | 254 nm (Optimized energy) | Standardized protein-RNA fixation. |
| Adapter Ligation | Single-stranded RNA ligase | Size-matched input (SMInput) control + Barcoded Adapters | Enables background subtraction; reduces PCR bias. |
| PCR Amplification | Variable cycles | Linear amplification via in-line barcodes & optimized cycles | Reduces duplicate reads, improves library complexity. |
| Validation | Often omitted | Mandatory Western blot post-ligation & pre-PCR | Confirms successful IP and library construction integrity. |
The most significant innovation is the inclusion of a size-matched input (SMInput) control. This control accounts for non-specific background arising from genomic DNA, abundant RNAs, and technical artifacts during RNA fragmentation and adapter ligation.
Detailed eCLIP Experimental Protocol
Cell Lysis and Crosslinked RNA Fragmentation
- Materials: UV-crosslinked cells (at 254 nm, ~150 mJ/cm²), IP Lysis Buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- Procedure: Lysate cells via sonication (Bioruptor) to shear DNA and reduce viscosity. Partial RNA fragmentation is achieved simultaneously. Treat lysate with RNase I (e.g., 1 µL of 1:1000 dilution per 10⁷ cells) to achieve fragments ~70-100 nucleotides. Clarify lysate by centrifugation.
Immunoprecipitation (IP) and Phosphatase/Kinase Treatment
- Materials: Pre-washed Protein A/G beads, validated antibody against target RBP, Phosphatase (FastAP), PNK (T4 Polynucleotide Kinase).
- Procedure: Incubate clarified lysate with antibody-bound beads overnight at 4°C. Wash stringently. On-bead, dephosphorylate RNA 3' ends with FastAP, then radiolabel RNA 5' ends with PNK and γ-³²P-ATP. Visualize successful IP via autoradiography after membrane transfer.
RNA Ligation, Protein Removal, and cDNA Synthesis
- Materials: T4 RNA Ligase 1, 3' pre-adenylated barcoded adapter, Proteinase K, Reverse Transcriptase (SuperScript IV), primer complementary to the ligated adapter.
- Procedure: Ligate a 3' adapter with unique molecular identifiers (UMIs) to the RNA on-bead. Elute RBP-RNA complexes and digest protein with Proteinase K. Extract RNA and ligate a 5' RNA adapter. Perform reverse transcription to generate cDNA.
cDNA Purification, PCR Amplification, and Sequencing
- Materials: Circligase, PAGE gel electrophoresis system, PCR reagents, Illumina sequencing adapters.
- Procedure: Circularize cDNA with Circligase and re-linearize with BamHI. Amplify libraries with a minimal number of PCR cycles (e.g., 8-15 cycles) using indexed primers. Size-select final libraries via gel purification (115-200 bp). Sequence on an Illumina platform (typically 50-100 million single-end reads per sample).
Achieving Single-Nucleotide Resolution Mapping
True single-nucleotide resolution is achieved by precisely mapping the crosslinking-induced mutation or truncation site.
- iCLIP/eCLIP Principle: The reverse transcriptase frequently terminates at the crosslinked nucleotide, creating cDNA truncations. Mapping these truncation sites identifies the crosslink position with nucleotide precision.
- PAR-CLIP Principle: Utilizes 4-thiouridine (4-SU) incorporation, leading to T-to-C transitions in sequencing reads. The site of this transition pinpoints the crosslink nucleotide.
- Data Analysis Pipeline: Key steps include:
- Demultiplexing & UMI Deduplication: Remove PCR duplicates using UMIs.
- Truncation Site Mapping: (For eCLIP/iCLIP) Identify the first nucleotide of the read (the cDNA truncation site) as the crosslink candidate.
- Mutation Site Mapping: (For PAR-CLIP) Identify significant T-to-C transitions above background.
- Peak Calling: Use tools like
CLIPperorPiranhaon the precise crosslink sites, comparing IP to SMInput control to define significant binding peaks.
Table 2: Quantitative Yield Metrics from a Representative eCLIP Study
| Sample Type | Final Library Concentration (nM) | PCR Cycles | % of Reads Aligning to Genome | % Duplicate Reads (Pre-UMI Dedup) | Significant Peaks Called |
|---|---|---|---|---|---|
| RBP IP | 12.5 | 14 | 85.2% | 65% | 12,450 |
| SMInput Control | 10.1 | 16 | 89.7% | 70% | 310 (background) |
Visualizing the eCLIP Workflow and Analysis
eCLIP Experimental Workflow with SMInput Control
eCLIP Data Analysis Pipeline to Single-Nucleotide Peaks
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for eCLIP and High-Resolution Mapping
| Item | Function in Protocol | Example/Specification |
|---|---|---|
| RNase I | Creates random, partial RNA fragments post-lysis, defining binding site resolution. | Thermo Fisher (EN0601); use at optimized dilution to yield ~70-100 nt fragments. |
| Pre-adenylated 3' Adapter | Ligation to RNA 3' end without ATP to prevent adapter concatemers. Contains UMI for deduplication. | IDT, 5'-rApp-NNNNNN...-3' (where N is UMI). |
| T4 RNA Ligase 1 (tr. K227Q) | High-efficiency ligase for single-stranded RNA adapters. Mutant version reduces ligation bias. | NEB (M0373). |
| Proteinase K | Digests the RBP after IP to release crosslinked RNA fragments for downstream library prep. | Roche (03115828001); used in strong buffer (SDS, EDTA). |
| SuperScript IV Reverse Transcriptase | Generates cDNA from crosslinked RNA with high processivity, improving yield of full-length cDNAs. | Thermo Fisher (18090010). |
| Circligase ssDNA Ligase | Circularizes single-stranded cDNA, a key step to prepare for PCR in iCLIP/eCLIP protocols. | Lucigen (CL4115K). |
| Validated RBP Antibody | Specific immunoprecipitation of the target RBP-RNA complex. Critical for success. | Recommend antibodies with published CLIP data (e.g., from Merck, Abcam). |
| Magnetic Protein A/G Beads | Solid support for antibody-based capture and washing of RBP complexes. | Pierce (88802/88803). |
Navigating Experimental Pitfalls: Troubleshooting and Optimization Strategies for CLIP-Seq
Within the context of CLIP-seq protocols (HITS-CLIP, PAR-CLIP, iCLIP), low yield is a critical bottleneck that can derail RBP-RNA interaction studies and subsequent drug discovery pipelines. This application note systematically diagnoses the three most common culprits: inadequate UV crosslinking, suboptimal antibody performance, and RNA degradation. We provide validated protocols and quantitative benchmarks to troubleshoot and optimize each step.
Quantitative Benchmarks for CLIP Yield Diagnostics
The following tables summarize expected values and failure indicators for key experimental checkpoints.
Table 1: Expected Yield Benchmarks Across CLIP Protocol Stages
| Protocol Stage | Expected Yield (HITS-CLIP) | Expected Yield (PAR-CLIP) | Critical Quality Check |
|---|---|---|---|
| RNase-Truncated RNA | 10-100 nucleotide fragments | 10-100 nucleotide fragments | Bioanalyzer/TapeStation profile |
| Immunoprecipitate (IP) RNA | 0.1-1% of input RNA | 0.1-1% of input RNA | qPCR for positive control RBP target |
| Final Library (Pre-seq) | 1-10 ng/µL in 10 µL | 1-10 ng/µL in 10 µL | Fragment size: ~150-300 bp |
| Adjusted cDNA (iCLIP) | 0.5-5 ng/µL | N/A | iCLIP-specific: cDNA smear on gel |
Table 2: Troubleshooting Matrix for Low Yield
| Symptom | Primary Suspect | Diagnostic Experiment | Confirmed if... |
|---|---|---|---|
| No RNA after IP | Antibody Performance | Western blot of IP pellet | No target RBP in pellet |
| Short RNA fragments (<10 nt) after IP | RNA Degradation | RNA Bioanalyzer (Pico chip) | Smear peaks at very low size |
| High background, low specific signal | Crosslinking Efficiency | Vary UV dose (e.g., 150-400 mJ/cm²) | Signal increases with optimized dose |
| No cDNA smear/ladder (iCLIP) | RNA Degradation / Ligation Efficiency | Test linker ligation on synthetic RNA | No ligation product observed |
Detailed Diagnostic Protocols
Protocol A: Assessing Crosslinking Efficiency
Purpose: To empirically determine the optimal UV dose for your RBP-cell system. Materials: UV crosslinker (254 nm), cell culture, ice-cold PBS. Procedure:
- Prepare six identical plates of cells at 70-80% confluence.
- Aspirate medium, wash cells gently with ice-cold PBS.
- Vary UV Dose: Expose each plate to a different UV dose (e.g., 0, 150, 200, 250, 300, 400 mJ/cm²) on ice.
- Proceed with standard cell lysis and RNA isolation.
- Perform a reverse crosslinking control: incubate an aliquot of lysate at 65°C for 1 hour to reverse inefficient crosslinks.
- Analyze RNA bound to your RBP via qPCR for a known binding site. The optimal dose yields the highest specific signal without increasing background (0 UV dose control).
Protocol B: Validating Antibody for CLIP
Purpose: To confirm antibody specificity and efficiency in native and crosslinked conditions. Materials: Target antibody, isotype control, Protein A/G beads, lysis buffer. Procedure:
- Prepare two lysates: one from UV-crosslinked cells and one from non-crosslinked cells.
- For each lysate, set up three IP reactions: a) Target antibody, b) Isotype control, c) No-antibody bead control.
- Incubate overnight at 4°C, wash stringently.
- Split each IP sample: Half for elution and Western blot (detects protein IP efficiency). Half for RNA isolation and qPCR (detects co-precipitated RNA).
- A valid CLIP antibody shows strong, specific protein pull-down and enriched RNA signal only in the UV-crosslinked + target antibody condition.
Protocol C: Monitoring RNA Integrity
Purpose: To detect and prevent RNase contamination at every stage. Materials: RNase inhibitors, RNA Pico Bioanalyzer chips, denaturing gels. Procedure:
- Pre-lysis Check: Spike a known amount of intact, exogenous RNA (e.g., in vitro transcript) into the lysis buffer. After lysis, re-purify it and run on a Bioanalyzer. Degradation indicates RNase in reagents.
- Post-RNase Truncation Check: After controlled RNase digestion, analyze 1% of the sample on a denaturing urea PAGE gel or Bioanalyzer. Expect a smear centered at ~50-70 nt. A sharp, very low molecular weight peak indicates over-digestion.
- Post-IP Wash Check: After final IP washes, resuspend beads in PK buffer, incubate at 55°C for 20 min, then purify RNA. The Bioanalyzer profile should mirror the post-RNase truncation profile.
Visualization of Diagnostic Workflows
Title: Diagnostic Decision Tree for Low CLIP Yield
Title: Core CLIP Experimental Workflow with Critical Steps
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for CLIP Diagnostics
| Item | Function & Importance in Diagnosis | Example/Notes |
|---|---|---|
| High-Fidelity UV Crosslinker | Ensures consistent, reproducible UV energy delivery for crosslinking efficiency tests. | UVP CL-1000 or equivalent with 254nm bulbs. Calibrate regularly. |
| RNase Inhibitor Cocktails | Prevents low-yield from RNA degradation during lengthy IP washes. | Use broad-spectrum, non-antibody based inhibitors (e.g., RNasin Plus). |
| Protein A/G Magnetic Beads | For efficient, low-background immunoprecipitation. Critical for antibody validation. | Bead choice depends on antibody species/isotype. |
| RNA Pico Bioanalyzer Chips | Provides precise, quantitative assessment of RNA fragment size and integrity post-IP. | Agilent 2100 Bioanalyzer system. Essential for Protocol C. |
| Phosphatase & Kinase Kits | For preparing RNA ends for adapter ligation. Inefficiency causes low library yield. | T4 PNK is standard. Check activity batches. |
| High-Specificity Antibody | The core reagent. Must be validated for IP under crosslinking conditions. | Use antibodies with published CLIP success or validate per Protocol B. |
| 4-Thiouridine (4SU) | For PAR-CLIP; increases crosslinking efficiency and introduces T-to-C mutations for site identification. | Optimize concentration for cell viability and incorporation. |
| Denaturing Urea-PAGE Gels | Gold-standard for visualizing size distribution of RNA fragments post-RNase digest. | More sensitive than Bioanalyzer for detecting small fragments. |
In UV crosslinking and immunoprecipitation (CLIP) methodologies—including HITS-CLIP, PAR-CLIP, and iCLIP—the precise mapping of RNA-protein interactions is paramount. A persistent challenge is the nonspecific background RNA signal that can obscure true binding sites. This background originates from non-crosslinked RNA co-purifying with the ribonucleoprotein (RNP) complex or from incomplete digestion of RNA fragments not protected by the bound RNA-binding protein (RBP). Effective management hinges on two critical, interdependent parameters: RNase concentration during fragmentation and wash stringency during immunoprecipitation (IP). This application note provides a systematic framework for optimizing these parameters to achieve high signal-to-noise ratios, framed within the broader thesis of refining CLIP-seq protocols for robust and reproducible RBP research in both basic and drug discovery contexts.
The Role of RNase and Wash Stringency in Noise Reduction
UV crosslinking covalently links an RBP to its bound RNA. Partial RNase digestion is then used to trim away unprotected RNA, leaving only a short "footprint" of the protein-protected region. Suboptimal RNase concentration leads to either over-digestion (loss of genuine signal) or under-digestion (increased background from unprotected RNA fragments). Following digestion and IP, stringent washing removes non-covalent associations. Insufficient stringency fails to remove nonspecifically bound RNA complexes, while excessive stringency may disrupt the antibody-antigen interaction or the crosslinked RNP itself.
Quantitative Optimization Data
Based on current literature and protocol benchmarks, the following tables provide guiding parameters. Optimal conditions must be empirically determined for each RBP and cell type.
Table 1: RNase A/T1 Concentration Ranges for CLIP Variants
| CLIP Protocol | Typical RNase A Range | Typical RNase T1 Range | Key Objective & Rationale |
|---|---|---|---|
| HITS-CLIP | 0.1 - 1.0 μg/mL | 0.01 - 0.5 U/μL | Generate ~50-70 nt footprints. Balances specificity with sufficient RNA for adapter ligation. |
| PAR-CLIP | 0.05 - 0.5 μg/mL | 0.005 - 0.1 U/μL | Milder digestion due to 4SU incorporation; aims for ~30-50 nt footprints to pinpoint crosslink sites. |
| iCLIP | 0.5 - 2.0 μg/mL (common: 1.0 μg/mL) | 0.1 - 1.0 U/μL (common: 0.25 U/μL) | More stringent digestion to produce very short fragments (<30 nt), reducing background for cDNA truncation mapping. |
| eCLIP | High-stringency Wash condition: 1:50 dilution of RNase I | Low-stringency Wash condition: 1:2000 dilution of RNase I | Uses RNase I. Systematic titration (e.g., 1:50, 1:200, 1:2000) is standard to identify optimal signal-to-noise. |
Table 2: Wash Buffer Stringency Comparison
| Wash Buffer Component | Low Stringency | Medium/Standard Stringency | High Stringency | Function |
|---|---|---|---|---|
| Salt (NaCl) | 150 mM | 300 - 500 mM | 1 M | Disrupts ionic interactions. Higher [NaCl] reduces nonspecific RNA-protein binding. |
| Detergent | 0.1% NP-40/Igepal | 0.1% - 0.5% SDS | 0.5% - 1% SDS | Disrupts hydrophobic interactions. SDS is highly denaturing. |
| Urea | 0 M | 0 M | 2 - 4 M | Chaotropic agent; denatures proteins and disrupts hydrogen bonding. |
| Lithium Chloride (LiCl) | 0 M | 0.25 M | 0.5 M | Alternative to NaCl; effective at removing nonspecific nucleic acids. |
| Typical Use Case | Pre-wash or for very labile complexes. | Common default for many RBPs (e.g., using High Salt Wash: 50 mM Tris-HCl, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate). | For RBPs with very high background or in optimized protocols like eCLIP (High Salt Wash followed by 2M Urea Wash). |
Detailed Experimental Protocols
Protocol 1: Empirical Titration of RNase Concentration
Objective: To determine the optimal RNase concentration that yields maximum unique cDNA reads from crosslinked RNA footprints while minimizing reads from longer, non-crosslinked RNA. Materials: Crosslinked cell lysate (pre-cleared), Anti-RBP antibody with beads, RNase A, RNase T1, Proteinase K, T4 PNK, SDS-PAGE reagents. Procedure:
- Prepare Lysate: Split 500 μL of pre-cleared lysate (from ~5x10^7 cells) into 5 x 100 μL aliquots.
- RNase Titration: Add RNase A/T1 mix to each aliquot to create a concentration series (e.g., Aliquot 1: 0.1 μg/mL RNase A + 0.01 U/μL RNase T1; Aliquot 2: 0.25+0.05; Aliquot 3: 0.5+0.1; Aliquot 4: 1.0+0.25; Aliquot 5: 2.0+0.5). Incubate at 22°C for 3-5 minutes.
- Immunoprecipitation: Add antibody-bound beads to each aliquot. Incubate at 4°C for 1-2 hours.
- Standardized Washes: Wash all samples identically with 3x of a medium-stringency wash buffer (e.g., 1X PBS, 0.1% SDS, 0.5% NP-40, 0.5% sodium deoxycholate).
- On-bead RNA Processing: Dephosphorylate with T4 PNK. Ligate 3' adapter. Radiolabel 5' ends with [γ-32P]ATP and T4 PNK.
- Visualization: Run samples on 4-12% Bis-Tris NuPAGE gel. Transfer to nitrocellulose, expose to phosphorimager. The optimal condition yields a clear, discrete RBP-RNA band shift with minimal smear at higher molecular weights.
- Validation: Proceed with full library prep from the optimal condition and sequence. Optimal condition maximizes the ratio of unique, mapping reads to repetitive/ribosomal reads.
Protocol 2: Systematic Optimization of Wash Stringency
Objective: To identify the wash stringency that maximally removes background RNA while retaining the specific RNP complex. Materials: Crosslinked, RNase-digested lysate (using predetermined optimal RNase conc.), Anti-RBP antibody with beads, Wash buffers of varying stringency. Procedure:
- Prepare Complexes: Incubate a large volume of digested lysate with antibody-bound beads. After binding, split the bead slurry into 4-5 equal aliquots.
- Differential Washing: Subject each aliquot to a different wash regimen:
- Set A: 3 x Low Stringency Buffer (e.g., 150 mM NaCl, 0.1% NP-40).
- Set B: 3 x Medium Stringency Buffer (e.g., 500 mM NaCl, 0.1% SDS, 0.5% NP-40).
- Set C: 1 x Medium Stringency, then 2 x High Salt Buffer (1 M NaCl, 0.1% SDS, 1% NP-40).
- Set D (eCLIP style): 2 x High Salt Buffer, then 2 x Urea Wash Buffer (2 M Urea, 20 mM Tris-HCl pH 7.5, 1% NP-40, 250 mM LiCl).
- Final Washes: All sets receive a final wash with 1X T4 PNK Buffer to prepare for downstream enzymatic steps.
- Analysis: Process all sets identically through on-bead kinase, adapter ligation, and autoradiography as in Protocol 1. The optimal wash condition yields the cleanest, most intense discrete band on the autoradiogram with the lowest high-molecular-weight smear. Sequence final libraries to confirm highest specificity (e.g., by peak enrichment over input).
Visualization of Workflow and Optimization Logic
Diagram 1: CLIP Noise Optimization Logic Flow
Diagram 2: Two-Phase Experimental Optimization Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Reagent | Function & Rationale in Noise Reduction |
|---|---|
| RNase A (heat-inactivated) | Endonuclease that cleaves after pyrimidines (C/U). Used in combination with T1 for general fragmentation. Heat inactivation removes DNase activity. |
| RNase T1 | Endonuclease specific for guanosine residues. Combined with RNase A for more uniform digestion patterns. Critical for producing short footprints. |
| RNase I | Non-specific endonuclease. Used in eCLIP for highly consistent digestion across all sequences. Requires careful titration. |
| Magnetic Protein A/G Beads | For immunoprecipitation. Superior wash efficiency over agarose beads, reducing nonspecific carryover. |
| Phosphatase & Kinase-Deficient T4 PNK Mutant | Used in iCLIP for 3' dephosphorylation without 5' kinase activity during intermediate steps, preventing aberrant labeling of background RNA. |
| 5'-3' Exonuclease (e.g., XRN1) | Used in some protocols to digest non-ligated RNA, reducing background from non-specific RNA fragments. |
| High-Salt Wash Buffers (e.g., with 1M NaCl) | Disrupts non-covalent ionic interactions between RNA and proteins or beads. Primary tool for increasing wash stringency. |
| Urea Wash Buffers (e.g., 2M Urea) | Chaotropic agent that denatures proteins, effectively removing proteins and RNA not covalently crosslinked to the target RBP. |
| SDS (Sodium Dodecyl Sulfate) | Ionic detergent that disrupts hydrophobic and electrostatic interactions. Powerful for eliminating nonspecific binding but must be balanced to maintain IP integrity. |
| 4-Thiouridine (4SU) | Nucleoside analog for PAR-CLIP. Induces T-to-C transitions upon crosslinking, providing a digital readout of binding sites and significantly reducing background during computational analysis. |
Within the broader thesis on CLIP-seq methodologies (HITS-CLIP, PAR-CLIP, iCLIP), a critical focus is the optimization of individual-nucleotide resolution crosslinking and immunoprecipitation (iCLIP). iCLIP provides unparalleled precision in identifying RNA-protein interaction sites by capturing crosslinked cDNA truncations at crosslink sites. However, its widespread adoption has been hampered by two persistent technical challenges: low yields of cDNA recovery after immunoprecipitation and inefficient circularization of the cDNA library prior to PCR amplification. These bottlenecks severely limit sequencing library complexity and overall experimental sensitivity. This application note details current, optimized protocols and reagent solutions to overcome these hurdles, enabling robust and reproducible iCLIP data generation for researchers and drug development professionals investigating RNA-binding proteins (RBPs).
The following table summarizes the typical yield losses at critical steps in standard iCLIP protocols and the improvement targets with optimized methods.
Table 1: Critical Yield Bottlenecks in Standard iCLIP Protocol and Optimization Goals
| Protocol Step | Typical Yield (Standard Protocol) | Primary Cause of Loss | Target Yield (Optimized) |
|---|---|---|---|
| RNA Adapter Ligation | 10-30% | Degraded adapter, inefficient T4 RNA Ligase | >70% |
| cDNA Synthesis & Recovery | 5-15% of input RNA | RNA degradation, inefficient reverse transcription, poor SPRI bead cleanup | 40-60% |
| cDNA Circularization | 10-25% | Inefficient Circligase, cDNA secondary structure | >80% |
| Final Library Amplification | Requires 18+ PCR cycles | Low input from preceding steps | Optimal: 12-15 cycles |
Optimized Experimental Protocols
Protocol 1: Enhanced cDNA Synthesis and Recovery
This protocol maximizes yield after reverse transcription (RT), the most critical point of loss.
Key Materials:
- RNA Sample: 3'-adapter ligated, crosslinked RNA-RBP complexes on beads.
- Reverse Transcriptase: Superscript IV (Thermo Fisher) or equivalent high-processivity, mutant RNase H- enzyme.
- RT Primer: iCLIP reverse transcription primer (5'-Phos/NNNNNnrGrGrG-3'; rG: RNA guanosine; n: random DNA base; N: molecular identifier).
- Cleanup Beads: RNase-free, size-selective SPRI beads (e.g., AMPure XP).
- Buffers: 5X Superscript IV buffer, 100 mM DTT, 10 mM dNTPs, RNaseOUT.
Detailed Procedure:
- Wash: Wash bead-bound complex twice with high-salt buffer (1M NaCl, 50mM Tris-HCl pH7.4) and once with 1X Superscript IV buffer.
- RT Master Mix: On ice, prepare:
- 8 µL 5X SSIV Buffer
- 2 µL 100mM DTT
- 2 µL 10mM dNTPs
- 2 µL RNaseOUT (80 U)
- 2 µL RT Primer (5 µM)
- 2 µL Nuclease-free H2O
- 2 µL Superscript IV (400 U)
- Reverse Transcription: Add 20 µL master mix to beads. Incubate:
- 2 min at 42°C
- 45 min at 52°C
- 15 min at 80°C (enzyme inactivation)
- Hold at 4°C.
- RNA Hydrolysis: Add 40 µL of RNase I/RNase A/T1 mix directly to RT reaction. Incubate 15 min at 37°C.
- cDNA Elution & Cleanup: Place on magnet, transfer supernatant containing cDNA to new tube. Add 1.8X volumes of SPRI beads, incubate 10 min, wash twice with 80% ethanol. Elute cDNA in 22 µL low TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0). Critical: Use a size-cut ratio (e.g., 0.8X to 1.8X double-sided cleanup) to remove excess primer and small fragments.
Protocol 2: High-Efficiency cDNA Circularization
This protocol addresses the inefficient intramolecular ligation of cDNA.
Key Materials:
- Circularization Enzyme: Circligase II ssDNA Ligase (Lucigen).
- Optimized Buffer: Use Circligase II buffer supplemented with 2.5 mM MnCl2 (freshly prepared).
- Betaine: 5M Betaine solution to disrupt secondary structure.
- cDNA: Purified cDNA from Protocol 1.
Detailed Procedure:
- Prepare Circularization Mix: In a PCR tube, combine:
- 20 µL purified cDNA (from Protocol 1, Step 5)
- 4 µL 10X Circligase II Buffer
- 4 µL 5M Betaine
- 2 µL 50 mM MnCl2
- 8 µL 50% PEG-6000 (optional but recommended)
- 1 µL Circligase II ssDNA Ligase (100 U)
- 1 µL Nuclease-free H2O
- Total Volume: 40 µL.
- Circularization Reaction: Incubate in a thermal cycler:
- 60 minutes at 60°C
- 10 minutes at 80°C (enzyme inactivation)
- Hold at 4°C.
- Cleanup: Add 1.8X volumes of SPRI beads to the reaction. Wash twice with 80% ethanol. Elute in 20 µL of 10 mM Tris-HCl, pH 8.0. The product is now ready for PCR amplification with iCLIP-specific primers.
Visualizing the Optimized iCLIP Workflow
Optimized iCLIP Workflow with Key Enhancements
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Overcoming iCLIP Hurdles
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Superscript IV RT | High-processivity, RNase H- mutant enzyme for superior cDNA yield from crosslinked, fragmented RNA. | Thermo Fisher, #18090010 |
| Circligase II ssDNA Ligase | Specialized thermostable ligase for efficient intramolecular circularization of single-stranded cDNA. | Lucigen, #CL9025K |
| RNA Cleanup Beads | Size-selective magnetic beads for precise removal of unligated adapters, primers, and small fragments. | Beckman Coulter, A63881 |
| Phosphorylated RT Primer | 5' phosphate group is essential for subsequent circularization step. | Custom DNA/RNA oligo synthesis. |
| Betaine (5M) | PCR additive that reduces secondary structure in cDNA, improving circularization efficiency. | Sigma-Aldrich, #B0300 |
| PEG 6000 | Macromolecular crowding agent to significantly boost ligation/ circularization rates. | Thermo Fisher, #AC327371000 |
| RNase I/T1 Mix | Efficiently degrades RNA post-RT without damaging the cDNA product. | Thermo Fisher, #AM2286 |
| Proteinase K | Essential for complete protein digestion after crosslinking reversal to release cDNA. | Roche, #03115828001 |
Within the broader landscape of CLIP-seq methodologies (HITS-CLIP, PAR-CLIP, iCLIP) for studying RNA-binding protein (RBP) dynamics, Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) offers superior crosslinking precision. It relies on the incorporation of nucleoside analogs like 4-thiouridine (4SU) into nascent RNA, followed by UV-A crosslinking. However, three interconnected challenges critically impact data quality and biological relevance: 4SU cytotoxicity, variable incorporation efficiency, and the consequent need for accurate mutation rate calibration during bioinformatic analysis.
Table 1: Impact of 4SU Concentration on Cell Viability and Incorporation
| 4SU Concentration (µM) | Treatment Duration (hr) | Relative Cell Viability (%) | 4SU Incorporation Level (RPKM) | Typical Use Case |
|---|---|---|---|---|
| 10-50 | 1-4 | 95-100 | Low | Short-term, sensitive cells |
| 100 | 4-16 | 80-90 | Moderate | Standard for many cell lines |
| 200-500 | 12-16 | 60-80 | High | Robust cell lines, high signal |
| >500 | >16 | <50 | Saturated but toxic | Generally not recommended |
Table 2: Key Bioinformatic Parameters for Mutation Calibration
| Parameter | Description | Typical Value/Threshold | Purpose |
|---|---|---|---|
| T-to-C Mutation Rate (Background) | Non-crosslinked, 4SU-containing RNA | 0.01-0.05% | Baseline noise estimation |
| T-to-C Mutation Rate (RBP-specific) | Crosslinked RNA from IP | 2-20% | Identifies true crosslink sites |
| Minimum Read Depth | At a candidate site | ≥ 20 reads | Statistical confidence |
| Mutation Enrichment Fold-Change | (IP T-to-C %) / (Background T-to-C %) | ≥ 5-10 fold | Filter for high-confidence sites |
Detailed Experimental Protocols
Protocol A: Titrating 4SU for Optimal Incorporation and Minimal Toxicity
Objective: Determine the optimal 4SU concentration that maximizes incorporation while maintaining >80% cell viability.
- Seed cells in multiple 6-well plates at 60% confluence.
- Prepare 4SU stock (e.g., 100 mM in DMSO) and dilute in complete medium to final concentrations (e.g., 10, 50, 100, 200, 500 µM). Include a DMSO-only control.
- Treat cells for a defined period (e.g., 16 hours).
- Assay Viability: Use trypan blue exclusion or a colorimetric assay (e.g., MTT) for one plate.
- Measure Incorporation: For parallel wells, extract total RNA. Perform cDNA synthesis and qPCR for housekeeping genes. Calculate relative incorporation via photoreversal assay (see Protocol B) or by quantifying mutations in sequenced rRNA.
- Select optimal concentration from the viability/incorporation curve.
Protocol B: Measuring 4SU Incorporation Efficiency via Photoreversal
- Split RNA sample (1 µg) from Protocol A into two aliquots.
- UV-A Irradiation: Expose one aliquot to 365 nm UV light (0.15 J/cm²) in a thin-walled PCR tube on ice. Keep the other in dark.
- cDNA Synthesis: Use reverse transcriptase (e.g., Superscript IV) with random hexamers on both samples.
- qPCR: Amplify a ~100-150 bp region of a highly expressed transcript (e.g., GAPDH, β-actin).
- Calculate: Incorporation efficiency is proportional to the difference in Ct values: ∆Ct = Ct(UV) - Ct(No UV). Higher ∆Ct indicates higher 4SU incorporation.
Protocol C: PAR-CLIP Library Prep with Mutation Calibration Controls
- Crosslink & Lysis: After 4SU incorporation, irradiate cells with UV-A (365 nm, 0.15 J/cm²). Lyse in stringent RIPA buffer.
- Immunoprecipitation: Use anti-target RBP antibody and protein A/G beads. Wash stringently.
- On-beads RNase T1 digestion to leave ~20-30 nt protein-protected RNA fragments.
- 3' Dephosphorylation (T4 PNK, minus ATP).
- 5' Phosphorylation & 3' Linker Ligation (T4 PNK with ATP; T4 RNA Ligase 1 with pre-adenylated 3' linker).
- Radioactive Labeling (Optional): Use [γ-³²P]ATP and T4 PNK to label RNAs for visualization.
- SDS-PAGE & Transfer: Isolate RBP-RNA complex, transfer to nitrocellulose, excise membrane slice corresponding to RBP's molecular weight.
- Proteinase K digestion to recover RNA.
- Reverse Transcription: Use a primer complementary to the 3' linker. Superscript III is recommended for its ability to read through crosslinked 4SU, introducing T-to-C mutations.
- cDNA Purification & 5' Linker Ligation.
- PCR Amplification with indexed primers. Include a background control library from input RNA (without IP) to assess baseline mutation rate.
- Sequencing (75-100 bp single-end reads recommended).
Visualization: Experimental Workflow and Logic
Diagram Title: PAR-CLIP Workflow with Key Challenges
Diagram Title: Bioinformatics Calibration for PAR-CLIP Mutations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for PAR-CLIP Experiments
| Reagent/Material | Function & Rationale | Key Consideration |
|---|---|---|
| 4-Thiouridine (4SU) | Nucleoside analog for UV-A crosslinking; induces T-to-C mutations. | Cytotoxicity requires titration. Use high-purity grade. |
| UV-A Lamp (365 nm) | Activates 4SU for crosslinking to RBPs. | Energy output must be calibrated (0.15-0.2 J/cm²) for consistency. |
| RNase T1 | Endoribonuclease for generating protein-protected RNA fragments. | Partial digestion is critical; optimize concentration. |
| Pre-adenylated 3' Linker | For ligation to RNA 3' ends without ATP to prevent circularization. | Essential for successful library construction from degraded RNA. |
| Proteinase K | Digests the RBP to recover crosslinked RNA fragments. | Must be molecular biology grade, free of RNases. |
| Superscript III Reverse Transcriptase | Reads through crosslinked 4SU, introducing diagnostic mutations. | Preferred over newer RTases for higher mutation incorporation. |
| Anti-RBP Antibody (High Quality) | Specific immunoprecipitation of the target RBP-RNA complex. | Validate for IP efficacy and specificity; crosslinking can alter epitopes. |
| Nitrocellulose Membrane | Captures RBP-RNA complexes after SDS-PAGE. | More efficient recovery of protein-nucleic acid complexes than PVDF. |
In the analysis of crosslinking and immunoprecipitation sequencing (CLIP-Seq) methods—including HITS-CLIP, PAR-CLIP, and iCLIP—data integrity is paramount for accurate identification of RNA-binding protein (RBP) interaction sites. PCR duplicates and adapter contamination constitute two critical "red flags" that can severely compromise downstream analyses, leading to false-positive peak calls and erroneous quantitative conclusions. This application note details their identification and mitigation within the specific context of RBP research, providing essential protocols for robust CLIP-Seq data processing.
Identifying and Quantifying PCR Duplicates
PCR duplicates are identical read pairs arising from the amplification of a single original DNA fragment. In CLIP-Seq, they can artificially inflate the signal at specific genomic locations.
Quantitative Indicators: A high percentage of PCR duplicates is a major red flag. Typical metrics are summarized below.
Table 1: Expected vs. Problematic PCR Duplicate Rates in CLIP-Seq
| CLIP Protocol | Expected Duplicate Rate | Red Flag Threshold | Primary Cause of High Duplicates |
|---|---|---|---|
| Standard RNA-Seq | 10-20% | > 30% | Low input material, over-amplification. |
| HITS-CLIP / iCLIP | 15-35% | > 50% | Very low starting RNA material due to crosslinking/cleavage. |
| PAR-CLIP | 20-40% | > 60% | Low input combined with T-to-C conversion sequencing. |
Protocol 1.1: In Silico Identification of PCR Duplicates
- Tool:
picard MarkDuplicatesorsamtools markdup. - Input: Coordinate-sorted BAM file from alignment.
- Method: Tools identify reads with identical 5' alignment coordinates (for unpaired data) or identical 5' coordinates for both mates (paired-end). One read is kept as "original," others are marked as duplicates.
- Command Example (Picard):
- Output: A BAM file with duplicate flags set and a metrics file detailing the percentage duplication.
Identifying and Quantifying Adapter Contamination
Adapter contamination occurs when sequencing reads contain portions of the Illumina adapter sequences, often due to short fragment sizes common in CLIP libraries. This can prevent alignment or cause misalignment.
Quantitative Indicators: The presence of adapter sequence is a clear sign of library preparation issues.
Table 2: Tools for Adapter Contamination Detection
| Tool | Method | Key Output Metric | Red Flag |
|---|---|---|---|
| FastQC | K-mer overrepresentation | Per-base sequence content plot showing enriched adapter sequences. | Sharp spikes in 'Overrepresented sequences' table. |
| cutadapt / Trim Galore! | Alignment of reads to adapter sequences | Percentage of reads containing adapter. | > 5-10% adapter content. |
| MultiQC | Aggregates FastQC/cutadapt reports | Summary across multiple samples. | Consistent adapter presence across samples. |
Protocol 2.1: Adapter Detection and Trimming
- Tool:
cutadapt - Input: Raw FASTQ files (R1 and R2 if paired-end).
- Method: The tool scans reads for partial or full adapter sequences and removes them.
- Command Example:
- Output: Trimmed FASTQ files and a report detailing the percentage of reads with adapters.
Integrated Workflow for CLIP-Seq QA/QC
A systematic preprocessing workflow is essential to mitigate these red flags before peak calling.
Title: CLIP-Seq Preprocessing & QA Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Robust CLIP-Seq Library Prep
| Reagent / Kit | Function in CLIP Protocols | Role in Mitigating Red Flags |
|---|---|---|
| RNase Inhibitor (e.g., SUPERase•In) | Protects RNA fragments during IP and library prep. | Preserves RNA integrity, reducing spurious fragments that lead to adapter dimers. |
| High-Sensitivity DNA/RNA Assay (e.g., Agilent Bioanalyzer/ TapeStation) | Accurate quantification and size profiling of libraries. | Identifies short fragment lengths (<100 bp) indicative of adapter contamination before sequencing. |
| Solid Phase Reversible Immobilization (SPRI) Beads | Size selection and clean-up post-ligation and PCR. | Critical for removing unligated adapters and primer dimers to prevent adapter contamination. |
| Reduced-Cycle PCR Kits (e.g., KAPA HiFi HotStart) | Amplification of cDNA library with high fidelity. | Minimizes PCR duplicate generation by using the fewest cycles necessary for sufficient yield. |
| Unique Molecular Identifiers (UMI) Adapters | Incorporation of random barcodes during adapter ligation. | Enables true duplicate removal at the molecular level, distinguishing PCR duplicates from biologically independent fragments. |
Protocol 3.1: UMI-Based Deduplication for iCLIP/HITS-CLIP
- Principle: UMIs are random nucleotide tags added to each molecule before PCR. True biological fragments with the same start site but different UMIs are retained.
- Tool:
UMI-toolsorfgbio. - Workflow:
- Extract UMIs: Parse UMI sequences from read headers or sequences.
- Deduplicate: Group reads by genomic coordinates and UMI, allowing for errors in UMI sequences.
- Command Example (UMI-tools):
- Output: A BAM file where reads with the same start position and UMI are collapsed, providing a true molecular count.
1. Introduction: Reproducibility in RBP-CLIP Studies Reproducibility is the cornerstone of rigorous RNA-binding protein (RBP) research using UV-crosslinking and immunoprecipitation (CLIP) techniques, including HITS-CLIP, PAR-CLIP, and iCLIP. Inherent technical variability from crosslinking efficiency, RNase digestion, library preparation, and bioinformatics analysis necessitates robust experimental design. This Application Note details the implementation of essential positive/negative controls and replicate strategies to ensure reliable, interpretable, and reproducible results in RBP-CLIP studies.
2. Essential Positive and Negative Controls: Definitions and Applications Controls are non-negotiable for validating both the experimental procedure and the specificity of the observed RNA-protein interactions.
Table 1: Mandatory Controls for CLIP-seq Experiments
| Control Type | Purpose | Recommended Implementation | Interpretation of Expected Result |
|---|---|---|---|
| Positive Technical Control (Input RNA) | Assess library preparation quality and background RNA abundance. | Use total RNA (no IP) from the same lysate. Sequence alongside CLIP libraries. | High correlation of abundant RNAs (e.g., ribosomal, mitochondrial) with CLIP sample indicates technical consistency. |
| Negative Technical Control (Beads-only / IgG) | Identify non-specific RNA binding to beads or antibody Fc region. | Perform IP with protein A/G beads + isotype control IgG (or beads alone) under identical conditions. | Minimal unique reads (<5-10% of target IP) indicates low background. Common contaminants (e.g., Malat1, Neat1) may appear. |
| Negative Biological Control (RNP Knockdown/Mutant) | Confirm RBP-specificity of binding sites. | Use siRNA/shRNA against target RBP, a catalytic/dead mutant, or a knockout cell line. Perform CLIP in parallel. | Significant reduction (>70%) in reads and peaks in knockdown vs. wild-type confirms specificity. |
| Spike-in Control (Synthetic RNA) | Normalize for technical variation cross-experiments. | Add known quantities of exogenous, non-crosslinkable RNAs (e.g., S. cerevisiae RNAs, ERCC Spike-Ins) to lysates. | Enables quantitative comparison of CLIP signal strength between different conditions or replicates. |
3. Experimental Protocol: Standardized iCLIP with Integrated Controls Protocol Title: Integrated Control iCLIP for Nuclear RBP A. Cell Culture & Crosslinking
- Grow two 15-cm plates of HeLa cells (WT and RBP-KO) to 80% confluency.
- Wash with PBS. For Positive Control: Harvest one well of WT cells for total RNA extraction (Input control).
- Irradiate plates with 254 nm UV-C light (400 mJ/cm²) on ice to crosslink RNA-protein complexes.
- Scrape cells into PBS, pellet, and flash-freeze.
B. Cell Lysis and Immunoprecipitation
- Lyse cell pellets in 1 mL of iCLIP lysis buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- For Beads-only Control: Reserve 100 µL of WT lysate. Add 50 µL pre-washed protein G magnetic beads. Incubate 1 hr at 4°C.
- For target IPs: Add 5 µg of specific anti-RBP antibody (or isotype control IgG for negative IP) to remaining lysates. Incubate 2 hrs at 4°C.
- Add 50 µL protein G beads to all IPs. Incubate 1 hr at 4°C.
- Wash beads 3x with high-salt wash buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate).
C. On-bead RNase Digestion, RNA Processing & Library Prep
- Critical: Perform limited RNase I digestion (diluted 1:100,000 in lysis buffer) for 3 minutes at 37°C to leave ~50-100 nt footprints.
- Dephosphorylate (T4 PNK, no ATP), ligate 3' RNA adapter.
- Radiolabel 5' ends with [γ-³²P]ATP using PNK. Visualize successful IP via autoradiography.
- Run complexes on NuPAGE gel. Transfer to nitrocellulose, isolate region above RBP size, and extract protein-RNA complexes.
- Digest protein with Proteinase K, purify RNA, reverse transcribe with barcoded primers.
- Perform cDNA circularization, PCR amplification, and size selection for sequencing.
4. Replicate Experiment Strategy and Statistical Power Biological replicates (independent cell cultures/experiments) are essential to distinguish consistent binding from stochastic noise. Technical replicates (multiple libraries from same IP) assess library prep variability.
Table 2: Replicate Design & Statistical Analysis Guidelines
| Replicate Type | Minimum Number | Purpose | Primary Analysis Metric |
|---|---|---|---|
| Biological | 3 (optimal), 2 (minimum) | Account for biological variability & enable statistical testing. | Irreproducible Discovery Rate (IDR) for peak calling; DESeq2 for differential binding. |
| Technical (Library Prep) | 2 | Assess amplification bias and sequencing noise. | Pearson correlation of read counts per gene/peak (R² > 0.95 expected). |
| Sequencing Depth | >10 million unique cDNA tags per biological replicate | Ensure saturation of detectable binding sites. | Plot cumulative novel peaks vs. sequencing depth. |
5. The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents for Reproducible CLIP
| Reagent / Material | Function | Critical Quality Check |
|---|---|---|
| High-Affinity, Validated Antibody | Specific immunoprecipitation of target RBP. | Validate by western blot and immunofluorescence in KO cell line. |
| RNase I (Ultrapure) | Controlled RNA fragmentation to generate precise footprints. | Titrate for each RBP to avoid over-/under-digestion. |
| Protein G Magnetic Beads | Efficient capture of antibody-RBP complexes. | Pre-clear with yeast tRNA to reduce non-specific RNA binding. |
| ³²P-γ-ATP or IRDye 800CW PNK | Visualization of successful IP and size selection. | Use fresh label; confirm signal on membrane before RNA extraction. |
| Barcoded Reverse Transcription Primers | Multiplexing of samples and reduction of index hopping effects. | Use unique dual indexes (UDIs) and purify via HPLC. |
| Spike-in RNA (e.g., ERCC, SIRV) | Normalization across conditions and batches. | Use a mix of non-crosslinkable RNAs added pre-IP. |
6. Visualizing Workflows and Logical Frameworks
Title: Integrated Control CLIP-seq Experimental Workflow
Title: Decision Tree for Validating CLIP-seq Peaks Using Controls
Benchmarking CLIP-Seq Data: Validation, Comparative Analysis, and Method Selection
Evaluating the success of CLIP-seq experiments (HITS-CLIP, PAR-CLIP, iCLIP) requires a multifaceted approach that assesses both technical quality and biological specificity. This application note provides a standardized framework of key metrics and detailed protocols for researchers studying RNA-binding proteins (RBPs) to ensure robust, reproducible data suitable for downstream analysis and drug discovery.
Within the broader thesis on CLIP-seq methodologies for RBP research, a critical challenge is the objective determination of data quality. High-throughput sequencing of crosslinked immunoprecipitation (CLIP) generates complex datasets where signal must be distinguished from noise. Success is not merely high read counts, but the specificity of RBP-RNA interaction capture. This document outlines the essential metrics and validation protocols to define experimental success.
Part 1: Key Quality Metrics and Quantitative Benchmarks
A successful CLIP-seq experiment is characterized by specific, reproducible enrichment of RNA targets. The following metrics, summarized in Table 1, should be calculated after standard preprocessing (adapter trimming, quality filtering) and alignment to the reference genome.
Table 1: Key CLIP-seq Quality Control Metrics and Benchmarks
| Metric Category | Specific Metric | Calculation/Description | Benchmark for Success | Implication of Poor Score |
|---|---|---|---|---|
| Library Complexity | Unique Deduplicated Reads | Reads remaining after PCR duplicate removal (using UMIs or positional deduplication). | > 1-5 million for mammalian RBPs. | High duplication indicates low starting material or over-amplification. |
| Complexity Ratio | (Deduplicated Reads) / (Total Mapped Reads). | > 0.2 - 0.5. | Ratios <0.1 suggest severe bottlenecking. | |
| Mapping & Signal | Genome Mapping Rate | (Reads mapped to genome) / (Total reads). | > 60-80%. | High unmapped rate may indicate contamination or adapter issues. |
| Reads in Peaks (RIP) | % of mapped reads falling within called crosslink sites. | 10-40% (varies by RBP). | <5% may indicate poor IP specificity or weak crosslinking. | |
| Signal-to-Noise | Signal-to-Background (Enrichment) | Fold-enrichment in IP over size-matched input (SMI) or IgG control at peak regions. | ≥ 4-fold enrichment. | Lack of enrichment indicates non-specific background. |
| Non-exonic Reads | % of reads mapping to intronic/intergenic regions. | Variable; often <30% for splicing regulators. | Abnormally high levels may indicate genomic DNA contamination. | |
| Crosslinking Specificity | Mutation Rate (PAR-CLIP) | % of T-to-C (for s4U) or C-to-T (for 6SG) transitions in read clusters. | ~5-20% at crosslink sites. | Low mutation rate suggests insufficient crosslink incorporation. |
| Deletion Rate (iCLIP) | % of reads with truncations at crosslink sites (cDNA deletions). | Significant enrichment at peak summits vs flank. | Absence suggests non-iCLIP background. | |
| Peak Characteristics | Peak Number | Significant peaks called (e.g., using PEAKachu, CLIPper, PyCRAC). | Hundreds to tens of thousands, biologically plausible. | Very few peaks indicate failed IP; excessive peaks may reflect noise. |
| Peak Width | Width of crosslink sites at base resolution. | iCLIP/HITS-CLIP: narrow (~1-3 nt). PAR-CLIP: slightly broader. | Broad, diffuse peaks suggest over-digestion or poor crosslinking. | |
| Reproducibility | Irreproducible Discovery Rate (IDR) | Consistency of peak calls between biological replicates. | IDR < 0.05 for high-confidence peaks. | High IDR indicates lack of reproducibility. |
Part 2: Detailed Protocol for Post-Sequencing Quality Assessment
This protocol details the computational steps to calculate the metrics in Table 1.
Protocol 2.1: Processing and Metric Calculation for CLIP-seq Data
I. Materials & Software (Research Reagent Solutions)
- Computational Environment: Linux/Unix server or high-performance computing cluster with ≥ 16GB RAM.
- Adapter Trimming: Cutadapt (v4.0+), fastp, or Trimmomatic.
- Alignment: STAR (v2.7+), Bowtie2, or HISAT2 with appropriate parameters for short, crosslinked reads.
- Deduplication: UMI-tools (for UMI-based protocols), Picard Tools'
MarkDuplicates, or custom scripts for positional deduplication. - Peak Calling: CLIPper (recommended for its specificity), PEAKachu, PyCRAC, or PureCLIP.
- Data Visualization: deepTools, Integrative Genomics Viewer (IGV), R/Bioconductor (ggplot2, ChIPseeker).
- Control Data: Size-Matched Input (SMI) or non-specific IgG control BAM file.
II. Procedure
Raw Data QC:
- Use FastQC to assess per-base sequence quality, adapter content, and nucleotide composition. Note any 3' bias or unusual k-mer profiles.
Adapter Trimming & Filtering:
cutadapt -a ADAPTER_SEQ -m 18 -j 8 -o output.trimmed.fastq.gz input.fastq.gz- Remove reads shorter than 18 nt (
-m 18) to avoid spurious alignments.
Alignment:
- Align to the reference genome plus splice junctions. Example with STAR:
STAR --genomeDir /path/to/genome --readFilesIn output.trimmed.fastq.gz --outFileNamePrefix sample1 --outSAMtype BAM SortedByCoordinate --runThreadN 8 --outFilterMultimapNmax 1 --alignEndsType Local - Set
--outFilterMultimapNmax 1to keep only uniquely mapped reads for most RBPs.
- Align to the reference genome plus splice junctions. Example with STAR:
Deduplication:
- For UMI protocols:
umi_tools dedup -I sample1.Aligned.sortedByCoord.out.bam --method unique -S sample1.dedup.bam - For non-UMI protocols: Use
samtools rmdupor Picard, noting this may over-deduplicate.
- For UMI protocols:
Generate Mapping Statistics:
- Use
samtools flagstaton the deduplicated BAM to calculate the genome mapping rate.
- Use
Peak Calling:
- Using CLIPper:
clipper -b sample1.dedup.bam -s hg38 --bonferroni --superlocal --threshold-method binomial -o sample1.peaks.bed - Use the
--bonferronicorrection for stringency. Provide a control BAM if available.
- Using CLIPper:
Calculate Key Metrics:
- Reads in Peaks (RIP): Use
bedtools intersectto count reads overlapping peaks. - Signal-to-Background: Compute read density (e.g., using
deepTools bamCoverage) in IP peaks versus the same regions in the control sample. - Mutation/Deletion Rates: For PAR-CLIP, use
PARalyzerorPyCRACto quantify T-to-C transitions. For iCLIP, analyze deletion profiles from the BAM files at peak coordinates. - Reproducibility: Call peaks on replicates independently, then use the
idrpackage to assess consistency.
- Reads in Peaks (RIP): Use
III. Interpretation
- Compile all calculated metrics into a summary table (as in Table 1).
- Successful experiments should pass benchmarks in most categories, particularly library complexity, enrichment over control, and reproducibility (IDR).
- Visual inspection in IGV is mandatory: peaks should show sharp, stranded signal in the IP, minimal signal in the control, and correspond to known RBP motifs or binding regions.
Part 3: Experimental Protocol for Wet-Lab Validation of Specificity
Computational metrics must be complemented by experimental validation.
Protocol 3.1: Crosslinking Efficiency and RNA Integrity Check (Pre-Sequencing)
I. Materials
- Radioisotope: γ-³²P-ATP.
- Reagents: T4 Polynucleotide Kinase (PNK), RNase Inhibitor, Proteinase K.
- Gel: 4-12% Bis-Tris NuPAGE gel.
- Membrane: Nitrocellulose.
- Apparatus: Semi-dry transfer system, Phosphorimager.
II. Procedure
- After UV crosslinking and cell lysis, split lysate.
- Treat one aliquot with Proteinase K to digest proteins and release crosslinked RNA.
- Radiolabel the RNA with PNK and γ-³²P-ATP.
- Run both treated (+PK) and untreated (-PK) samples on a denaturing NuPAGE gel.
- Transfer to nitrocellulose. The membrane retains proteins and protein-RNA complexes.
- Visualize by phosphorimaging.
III. Expected Results & Metric
- Success: A radioactive smear on the membrane in the -PK lane, shifting lower (smaller RNA fragments) in the +PK lane. This confirms RNA was successfully crosslinked to the RBP.
- Metric: The efficiency can be approximated by the fraction of radiolabeled RNA retained on the membrane in the -PK lane versus migrating through in the +PK lane. >30% retention is typical.
Part 4: Visualizing Workflows and Relationships
Title: Computational CLIP-seq Quality Assessment Workflow
Title: Sources of Specific Signal vs. Background in CLIP
Part 5: The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for CLIP-seq Quality Control
| Reagent/Tool Category | Specific Name/Type | Function in Quality Assessment |
|---|---|---|
| Crosslinking & Lysis | UV-C Lamp (254 nm), 4-Thiouridine (s4U) / 6-Thioguanosine (6SG) | Induces covalent protein-RNA bonds. Nucleotide analogs enable specific mutation signatures for PAR-CLIP. |
| Immunoprecipitation | High-Specificity Anti-RBP Antibody (validated for CLIP), Magnetic Protein A/G Beads | Enriches for RBP-RNA complexes. Antibody specificity is the single most critical wet-lab factor. |
| Library Prep | Unique Molecular Identifiers (UMIs), RNase Inhibitors, High-Fidelity Polymerase | UMIs enable accurate PCR duplicate removal. Inhibitors maintain RNA integrity. |
| Validation | γ-³²P-ATP, T4 PNK, Proteinase K, Nitrocellulose Membrane | For pre-sequencing radiometric assay to check crosslinking efficiency and RNA integrity. |
| Computational | Cutadapt/Trimmomatic, STAR/Bowtie2, UMI-tools, CLIPper, deepTools, IDR | Software pipeline for read processing, alignment, deduplication, peak calling, and metric calculation. |
| Control Samples | Size-Matched Input (SMI), Non-specific IgG IP, Knockout/Knockdown Cell Line | Essential for distinguishing true signal from technical and biological background. |
Within the broader thesis on CLIP-seq methodologies for RNA-binding protein (RBP) research, selecting the appropriate protocol is critical. HITS-CLIP, PAR-CLIP, and iCLIP each offer distinct advantages and trade-offs in resolution, signal-to-noise ratio, and technical complexity. This application note provides a head-to-head comparison to guide researchers and drug development professionals in protocol selection based on specific project goals.
Quantitative Comparison Table
Table 1: Core Characteristics of CLIP-seq Variants
| Feature | HITS-CLIP (CLIP-seq) | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslinking Method | UV-C (254 nm) | UV-B (365 nm) + 4-Thiouridine (4SU) / 6-Thioguanosine (6SG) | UV-C (254 nm) |
| Key Mutational Signal | Deletions (at crosslink site) | T-to-C (4SU) or G-to-A (6SG) transitions | cDNA truncations (at crosslink site +1) |
| Theoretical Resolution | ~30-60 nt (protein footprint) | Nucleotide-level (via mutation) | Nucleotide-level (via truncation) |
| Signal-to-Noise Ratio | Moderate | High (reduced background from mutation counting) | High (unique truncation signal) |
| Primary Technical Demand | Moderate | High (requires metabolic labeling & specialized sequencing analysis) | High (complex library prep, precise adapter ligation) |
| RBP Applicability | Broad | Requires cellular 4SU/6SG incorporation; less suitable for in vivo/tissue | Broad, excellent for RBPs with dense or overlapping binding |
| Key Advantage | Robust, widely established protocol | Highest precision in defining binding sites | Unlocks protein-RNA interaction sites & structural insights |
Table 2: Typical Sequencing & Analytical Metrics
| Metric | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Recommended Sequencing Depth | 10-20 million reads | 10-30 million reads | 20-40 million reads |
| Mapping Rate | ~70-85% | Lower (~50-70%) due to mutations | ~60-80% |
| Primary Analysis Software | CLIPper, Piranha | PARalyzer, wavClusteR | iCount, iMaps, PureCLIP |
| Mutation/Truncation Rate at Sites | Low deletion rate | High (>5% T-to-C) | Variable truncation rate |
Detailed Experimental Protocols
Protocol 1: Core Crosslinking & Immunoprecipitation (Shared Initial Steps)
- Cell Preparation: Grow adherent or suspension cells to ~80% confluence.
- Metabolic Labeling (PAR-CLIP only): Supplement growth medium with 100 µM 4-Thiouridine (4SU) for one cell doubling period (~16 hrs).
- UV Crosslinking:
- HITS-CLIP & iCLIP: Wash cells with ice-cold PBS. Irradiate once with 254 nm UV-C at 150-400 mJ/cm² in a Stratalinker. Perform on ice.
- PAR-CLIP: Wash 4SU-labeled cells. Irradiate with 365 nm UV-B at 0.15-0.45 J/cm².
- Cell Lysis: Scrape cells in lysis buffer (e.g., 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, protease/RNase inhibitors).
- Partial RNase Digestion: Add RNase I (e.g., 0.01-0.1 U/µL) to digest RNA and leave ~20-70 nt protein-protected footprints. Incubate 3-15 min at 22°C or 37°C.
- Immunoprecipitation: Pre-clear lysate. Add antibody against target RBP (or epitope tag) and incubate with Protein A/G magnetic beads for 1-2 hours at 4°C.
- Washing: Wash beads stringently with high-salt buffer (e.g., 50 mM Tris-HCl, 1M NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) and low-salt buffer.
Protocol 2: iCLIP-Specific Adapter Ligation & Library Prep
This protocol highlights the key divergent step for iCLIP.
- 3' Dephosphorylation: After washing, treat beads with T4 PNK (without ATP) in PNK buffer to dephosphorylate RNA 3' ends. Wash.
- 3' Adapter Ligation: Ligate a pre-adenylated DNA adapter (e.g., /5rApp/AGATCGGAAGAGCGGTTCAG/3ddC/) to the RNA 3' ends using T4 RNA Ligase 1 (truncated) in a buffer without ATP. Incubate overnight at 16°C.
- Radiolabeling (for visualization): Transfer beads to a new tube. Perform 5' phosphorylation with PNK and [γ-³²P]ATP. Run on NuPAGE gel, transfer to nitrocellulose membrane, and expose to film to isolate the correct RBP-RNA complex.
- Proteinase K Digestion: Excise membrane region above the RBP's size. Digest with Proteinase K to release RNA.
- Reverse Transcription (RT): Purify RNA. Perform RT with a primer complementary to the 3' adapter. The RT will frequently terminate at the crosslinked nucleotide (+1).
- cDNA Circularization & PCR: Circularize cDNA using Circligase. Re-linearize and amplify with PCR primers containing Illumina adapters and sample indexes.
Protocol 3: PAR-CLIP-Specific Sequencing Library Construction
This protocol highlights mutation detection.
- 3' Adapter Ligation (On-bead): After washing, directly ligate a pre-adenylated DNA 3' adapter using T4 RNA Ligase 1 (truncated).
- 5' Phosphorylation & Radiolabeling: As in iCLIP Protocol Step 3, use PNK to radiolabel and visualize the complex, followed by membrane excision and proteinase K digestion.
- RNA Purification & 5' Adapter Ligation: Purify RNA. Ligate an RNA 5' adapter using T4 RNA Ligase 1.
- Reverse Transcription and PCR: Perform RT with a primer against the 3' adapter, followed by PCR with Illumina-compatible primers.
- Sequencing & Mutation Analysis: Sequence. Use tools like PARalyzer to identify clusters enriched for T-to-C (or G-to-A) transitions, defining crosslink sites.
Visualization of Workflows & Concepts
CLIP-seq Method Selection Workflow
Decision Tree for CLIP Method Selection
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for CLIP-seq
| Reagent / Solution | Function & Critical Notes |
|---|---|
| 4-Thiouridine (4SU) | Photoreactive nucleoside for PAR-CLIP. Incorporated into nascent RNA, enables efficient crosslink under 365 nm UV and induces T-to-C mutations. |
| RNase I | Endoribonuclease for partial RNA digestion. Concentration is critical to generate optimal protein-protected RNA footprints (20-70 nt). |
| Protein A/G Magnetic Beads | For immunoprecipitation of antibody-RBP-RNA complexes. Magnetic beads facilitate stringent washing. |
| Pre-adenylated 3' Adapter | Essential for iCLIP (and used in others). The 5' adenylation (/5rApp/) and 3' ddC block enable ligation only to RNA 3' ends without ATP, reducing adapter dimer formation. |
| T4 RNA Ligase 1 (Truncated) | Specifically used for ligating pre-adenylated adapters to RNA 3' ends. Lacks the activity that promotes circularization or multimer formation. |
| T4 Polynucleotide Kinase (PNK) | Used for 5' end radiolabeling (with [γ-³²P]ATP) for complex visualization and for 3' dephosphorylation in iCLIP. |
| Proteinase K | Digests the RBP after membrane excision to recover crosslinked RNA fragments for library construction. |
| NuPAGE Bis-Tris Gels & Nitrocellulose Membrane | For size separation and transfer of radiolabeled RBP-RNA complexes. Allows precise excision of the target complex to reduce background. |
| PARalyzer / wavClusteR Software | Specialized algorithms for identifying significant crosslink sites from PAR-CLIP data by statistically modeling mutation rates. |
| iCount / PureCLIP Software | Analysis tools designed to identify significant crosslink sites from iCLIP and other CLIP data, focusing on cDNA truncation events. |
Within the landscape of RNA-binding protein (RBP) research, high-throughput CLIP-seq protocols (HITS-CLIP, PAR-CLIP, iCLIP) are foundational for mapping in vivo RBP-RNA interactions. However, the biological interpretation of these maps requires orthogonal validation to distinguish direct from indirect binding, quantify affinity, and determine functional consequences. This application note details a framework integrating transcriptome-wide assays (RIP-seq, RNA-seq) with in vitro biophysical assays (RBNS, MITOMI) to create a multi-layered, validated model of RBP function, directly supporting and refining hypotheses generated from primary CLIP-seq data.
Table 1: Orthogonal Assays for CLIP-seq Validation
| Assay | Key Measurement | Throughput | Context | Primary Validation Role |
|---|---|---|---|---|
| CLIP-seq (e.g., iCLIP) | In vivo binding sites, nucleotide resolution | Genome-wide | Native cellular environment | Primary discovery of binding landscapes. |
| RIP-seq | Transcriptome-wide RNA association | Genome-wide | Native, but less stringent | Confirms in vivo association; lower resolution. |
| RNA-seq | Transcript abundance & alteration | Genome-wide | Native cellular environment | Identifies functional consequences (splicing, stability). |
| RBNS | Relative binding affinity & motif discovery | Medium-high (Oligo libraries) | Purified RBP, in vitro | Quantifies sequence affinity; defines core motif. |
| MITOMI | Absolute binding kinetics (K_d) | Low-medium (Designed sequences) | Purified RBP, in vitro | Measures precise kinetic & equilibrium constants. |
The integration strategy follows a convergent logic: 1) RIP-seq confirms in vivo association of targets identified by CLIP-seq. 2) RNA-seq on RBP perturbation (knockdown/overexpression) links binding to functional change. 3) RBNS delineates the intrinsic sequence preference from the cellular CLIP-seq map. 4) MITOMI provides quantitative biophysical validation for top candidate motifs.
Detailed Experimental Protocols
Protocol 3.1: RIP-seq for In Vivo Association Validation
Objective: To validate CLIP-seq-identified RNA targets via immunoprecipitation under native conditions. Materials: Cell line expressing RBP of interest, RIP lysis buffer (150mM KCl, 25mM Tris pH 7.4, 5mM EDTA, 0.5% NP-40, protease/RNase inhibitors), Protein A/G magnetic beads, validated antibody (vs. RBP or epitope tag), TRIzol. Procedure:
- Lyse 1x10^7 cells in 1 ml RIP lysis buffer (10 min, 4°C).
- Clear lysate by centrifugation (14,000g, 10 min, 4°C).
- Incubate 10% input aliquot with TRIzol (-80°C store). Incubate remainder with 5 µg antibody-conjugated beads (1 hr, 4°C).
- Wash beads 5x with lysis buffer.
- Isolate RNA from beads and input aliquot using TRIzol/chloroform.
- Generate sequencing library (ribodepletion recommended) from both IP and input RNA. Sequence.
- Analyze: Enriched transcripts in IP vs. input (tools: Salmon/DESeq2) are compared to CLIP-seq target list.
Protocol 3.2: RNA-seq for Functional Consequence
Objective: To identify RBP-mediated changes in RNA processing/abundance. Materials: Cells with RBP knockdown (siRNA) vs. control, TRIzol, poly(A) selection or ribodepletion kits, strand-specific library prep kit. Procedure:
- Generate biological triplicates of knockdown (KD) and control cells.
- Extract total RNA with TRIzol, assess quality (RIN > 8).
- Prepare strand-specific RNA-seq libraries (polyA-selected for mRNA; ribodepleted for total RNA).
- Sequence to a depth of 30-40 million paired-end reads per sample.
- Analyze: Map reads (STAR), quantify transcripts (StringTie), perform differential expression/alternative splicing analysis (DESeq2, DEXSeq, rMATS). Integrate differentially expressed/spliced genes with CLIP-seq/RIP-seq binding targets.
Protocol 3.3: RBNS (RNA Bind-n-Seq) for Affinity Motif Discovery
Objective: To determine the in vitro sequence/structural preference of the purified RBP. Materials: Purified recombinant RBP (≥95% pure), NGS-based randomized RNA oligo library (e.g., 40nt random region), nitrocellulose filter membrane (0.45 µm) or EMSA gel, library prep kit. Procedure:
- Binding Reactions: Incubate fixed RBP concentration (nM range) with randomized library (1nM) in binding buffer across a range of increasing competitor (polyrA, polyrU, tRNA) concentrations.
- Partitioning: Pass reactions through nitrocellulose filter (retains protein-bound RNA). Elute bound RNA. Alternative: native gel extraction.
- Amplification & Sequencing: Reverse transcribe eluted RNA, PCR amplify, and prepare for high-throughput sequencing.
- Analysis: Align reads, count enrichment of k-mers at each selection round/competitor level relative to input library. Tools: k-mer counting scripts, MEME for motif discovery. Compare RBNS-derived motif to CLIP-seq crosslink motif.
Protocol 3.4: MITOMI (Mechanically Induced Trapping of Molecular Interactions) for Kinetics
Objective: To measure precise dissociation constants (K_d) for specific RBP-RNA interactions. Materials: MITOMI microfluidic device, purified RBP (fluorescently tagged), synthetic RNA targets (Cy5-labeled), PBS-T buffer. Procedure:
- Device Priming: Fabricate PDMS microfluidic device with button valves. Functionalize flow chambers with anti-His/Strep antibody to capture His/Strep-tagged RBP.
- RBP Loading: Flow fluorescent RBP into device, capture in designated chambers.
- Binding Assay: Flow a gradient of concentrations of Cy5-labeled RNA (0.1nM - 1µM) over captured RBP. Allow equilibrium.
- Trapping: Activate "button" valve to mechanically trap complexes against the surface.
- Washing & Imaging: Wash unbound RNA away, image fluorescence (RBP channel, RNA channel) for each concentration.
- Analysis: Quantify fluorescence intensities. Fit RNA signal vs. concentration to a 1:1 binding isotherm to extract K_d.
Visualizing the Orthogonal Validation Workflow
Diagram Title: Orthogonal Validation Workflow for RBP Research
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 2: Essential Reagents for Orthogonal RBP Studies
| Reagent/Solution | Function | Example/Notes |
|---|---|---|
| UV Crosslinker (254 nm) | Covalently freeze in vivo RBP-RNA interactions for CLIP-seq. | Critical for standard CLIP protocols. PAR-CLIP uses 4-thiouridine & 365 nm. |
| RNase Inhibitors | Preserve RNA integrity during immunoprecipitation & lysis. | Use broad-spectrum inhibitors (e.g., RNasin, SUPERase•In). |
| Magnetic Beads (Protein A/G) | Solid-phase support for antibody-based RIP. | Enable efficient washing; reduce background. |
| High-Affinity RBP Antibodies | Specific immunoprecipitation of native RBP. | Validate for IP-grade. Epitope tags (FLAG, HA) offer an alternative. |
| Randomized RNA Oligo Library | Defined, diverse pool for in vitro selection (RBNS). | Typically 10^12 unique sequences with central random region. |
| Recombinant RBP (Purified) | Source of protein for in vitro assays (RBNS, MITOMI). | Requires high purity (>95%) and confirmed RNA-binding activity. |
| Cy5/Cy3-labeled RNA Oligos | Fluorescent probes for quantitative binding (MITOMI, EMSA). | HPLC-purified; used for K_d determination. |
| Strand-Specific RNA-seq Kit | Preserves directionality of transcripts for functional analysis. | Critical for identifying antisense transcription & precise TSS. |
| Microfluidic MITOMI Device | Miniaturized platform for parallel binding kinetics measurement. | Enables high-precision, multi-condition K_d measurement. |
This protocol details the essential bioinformatics validation pipeline for CLIP-seq variants (HITS-CLIP, PAR-CLIP, iCLIP) used in RNA-binding protein (RBP) research. Within the broader thesis on experimental RBPs research, this computational workflow is critical for transforming raw sequencing data into biologically interpretable results, linking RBP binding sites to molecular function and potential drug targets.
Application Notes & Protocols
Peak Calling: Identifying Significant RBP Binding Sites
Peak calling distinguishes true protein-RNA binding events from background noise. The choice of tool depends on the CLIP-seq protocol.
Protocol: Peak Calling with PEAKachu (for iCLIP/eCLIP data)
- Input Preparation: Convert demultiplexed BAM files (aligned reads) to BED format using
bedtools bamtobed. - Background Model: Generate a matched input or shuffled background control BED file.
- Tool Execution: Run PEAKachu in differential mode for robust calling.
- Post-processing: Filter peaks based on p-value (e.g.,
p < 0.05) and fold-enrichment over control (e.g.,FC > 2). Merge adjacent peaks within 50 nucleotides usingbedtools merge.
Table 1: Comparison of Peak Calling Tools for CLIP-seq
| Tool | Best Suited For | Key Metric | Recommended Cut-off | Primary Output |
|---|---|---|---|---|
| PEAKachu | iCLIP, eCLIP | Statistical score (p-value) | p < 0.01 | BED file of peaks |
| PARalyzer | PAR-CLIP | T-to-C mutation density | Read count > 10 | GRanges (R) / BED |
| PIPE-CLIP | HITS-CLIP, mixed protocols | Peak height & SNR | FDR < 0.05 | BED file of peaks |
De novo motif analysis identifies over-represented sequence or structural patterns within called peaks.
Protocol: De Novo Motif Finding with MEME-ChIP
- Sequence Extraction: Using
bedtools getfasta, extract genomic sequences underlying high-confidence peaks (e.g., top 500 by p-value), plus 20-nt flanks. - MEME Execution: Run the MEME-ChIP suite for comprehensive discovery.
- Analysis: Examine
meme-chip.htmloutput. Primary results include:- MEME: De novo motif position weight matrices (PWMs).
- CentriMo: Verification of motif centrality in peaks.
- Tomtom: Comparison to known RBP motifs in databases (e.g., CISBP-RNA, ATtRACT).
Table 2: Representative Motif Discovery Results for an RBP (Example)
| Discovered Motif (Sequence Logo) | E-value (Significance) | Best Match to Known RBP (Tomtom q-value) | Genomic Context (CentriMo p-value) |
|---|---|---|---|
| UGCAUGU | 1.2e-10 | FOX2 (q=0.002) | Significant central enrichment (p<0.001) |
| UAUUUAU | 5.7e-08 | ELAVL1 (HuR) (q=0.015) | Moderate central enrichment (p<0.05) |
Functional Enrichment Analysis: From Binding Sites to Biology
This step links RBP binding targets to cellular pathways, functions, and diseases.
Protocol: Integrated Enrichment using g:Profiler & clusterProfiler
- Gene Annotation: Map peak locations to genes using
ChIPseeker(R/Bioconductor). Define gene as "target" if a peak falls within its transcript, 3' UTR, or 5' UTR. - Gene List Enrichment: Submit the target gene list to the g:Profiler web interface (or use
gprofiler2R package) for functional terms (GO, KEGG, Reactome). - Visualization & Over-Representation Analysis (ORA): In R, use
clusterProfilerfor detailed visualization and statistical testing.
Table 3: Example Functional Enrichment Results (Top 5 KEGG Pathways)
| Pathway Name | Gene Ratio (Target/Total) | Adjusted p-value | Candidate Drug Targets in Pathway |
|---|---|---|---|
| mRNA surveillance pathway | 12/169 | 3.5E-08 | SMG1, UPF1 |
| RNA transport | 18/170 | 1.2E-06 | XPO1, EIF4E |
| Spliceosome | 15/130 | 4.7E-05 | SF3B1, PRPF8 |
| Autophagy | 9/150 | 0.002 | MTOR, BECN1 |
| Neurotrophin signaling pathway | 8/120 | 0.012 | NTRK1, MAPK1 |
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in CLIP-seq Bioinformatics Pipeline |
|---|---|
| R/Bioconductor Packages (ChIPseeker, GenomicRanges) | For peak annotation, genomic interval manipulation, and statistical analysis in R. |
| Conda/Bioconda Environment | For reproducible installation and management of bioinformatics software versions. |
| ATtRACT or CISBP-RNA Motif Database | Curated databases of known RBP binding motifs for motif comparison (Tomtom). |
| g:Profiler Web Service / API | Fast, integrated functional enrichment analysis across multiple annotation databases. |
| UCSC Genome Browser Session | Visual validation of called peaks in genomic context against public tracks (e.g., conservation, splicing). |
| High-Performance Computing (HPC) Cluster Access | Essential for processing large CLIP-seq datasets through compute-intensive alignment and peak calling steps. |
Visualizations
Diagram 1: CLIP-seq Bioinformatics Validation Workflow
Diagram 2: From Peak to Pathway Logic
RNA-binding proteins (RBPs) are central regulators of post-transcriptional gene expression. Mapping their precise interactions with RNA is critical for understanding cellular function and dysfunction. This application note, framed within a thesis on CLIP-Seq variants, provides a decision matrix and detailed protocols to guide researchers in selecting the optimal method—HITS-CLIP, PAR-CLIP, or iCLIP—based on RBP properties, cell type, and specific research goals.
Protocol Decision Matrix
The choice of Crosslinking and Immunoprecipitation (CLIP) protocol depends on several interconnected factors. The following matrix summarizes the key decision criteria.
Table 1: CLIP-Seq Protocol Selection Matrix
| Criterion | HITS-CLIP | PAR-CLIP | iCLIP |
|---|---|---|---|
| Crosslink Type & Resolution | UV-C (254 nm) induces protein-RNA crosslinks via protein-nucleic acid interactions. Resolution: ~1 nucleotide. | UV-C (365 nm) + 4-thiouridine (4SU) induces T-to-C transitions. Resolution: ~20-30 nucleotides. | UV-C (254 nm). cDNA truncation at crosslink site provides single-nucleotide resolution. |
| Optimal RBP Properties | Robust crosslinker; suitable for most RBPs, especially those binding pre-mRNA or with lower crosslinking efficiency. | Requires cellular incorporation of 4SU. Ideal for studying RBPs in systems with high RNA turnover (e.g., cultured cells). | Excellent for studying RBPs with tight binding or that crosslink at low efficiency. Ideal for mapping precise binding boundaries. |
| Cell Type Considerations | Versatile: tissues, primary cells, in vivo models, neurons. No nucleotide analog required. | Best for cell cultures (mammalian, yeast) amenable to 4SU labeling. Less suitable for in vivo/tissue samples. | Versatile like HITS-CLIP. Particularly powerful for complex tissues and in vivo contexts. |
| Primary Research Goal | Genome-wide binding map identification, alternative splicing analysis. | Highest signal-to-noise ratio; precise binding site identification via mutation signature. | Single-nucleotide resolution mapping, studying protein-RNA interactions that block reverse transcription. |
| Key Advantage | Broad applicability, no need for metabolic labeling. | Reduced background, diagnostic mutations pinpoint crosslink sites. | Identifies exact crosslink site via cDNA truncation, reveals structural insights. |
| Key Limitation | Higher background noise, precise crosslink site inference is indirect. | Requires 4SU incorporation, which can be toxic and alter cellular physiology. | Technically more challenging, lower library complexity requires higher sequencing depth. |
Detailed Experimental Protocols
Protocol 1: iCLIP (Individual-nucleotide resolution CLIP)
Principle: UV crosslinking, stringent immunoprecipitation, and a circularization-based library preparation that captures cDNAs truncated at the crosslink site.
Materials & Reagents:
- UV-C Crosslinker (254 nm)
- IP Wash Buffer: 50 mM Tris-HCl (pH 7.4), 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate.
- PNK Buffer: 70 mM Tris-HCl (pH 7.6), 10 mM MgCl₂, 5 mM DTT.
- T4 PNK (NEB): For 3' dephosphorylation and 5' phosphorylation.
- 3' RNA Adaptor (L3-App): Pre-adenylated for ligation.
- SDS-PAGE & Transfer Equipment: For protein-RNA complex separation.
- Nitrocellulose Membrane: For transfer and complex immobilization.
- Proteinase K Buffer: 100 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM EDTA, 0.2% SDS.
- CircLigase II (Lucigen): For single-stranded DNA circularization.
- High-Fidelity PCR Enzyme (e.g., KAPA HiFi): For final library amplification.
Procedure:
- Crosslinking & Lysis: Irradiate cells/tissue with 254 nm UV-C (150-400 mJ/cm²). Lyse in stringent RIPA buffer.
- Partial RNase Digestion: Treat lysate with limited RNase I to generate RNA footprints (~50-70 nt).
- Immunoprecipitation: Incubate with antibody-bound beads. Wash stringently with IP wash buffer.
- 3' Dephosphorylation & 5' Phosphorylation: On beads, treat with T4 PNK in PNK buffer to prepare for adaptor ligation.
- 3' RNA Adaptor Ligation: Ligate pre-adenylated 3' adaptor (L3-App) using T4 RNA Ligase 2, truncated.
- Complex Isolation: Run sample on SDS-PAGE, transfer to nitrocellulose membrane, and excise region above the IgG heavy chain.
- Proteinase K Digestion: Elute and digest complexes with Proteinase K to recover RNA.
- Reverse Transcription: Use a primer complementary to the 3' adaptor. Reverse transcriptase will truncate at the crosslink site.
- cDNA Circularization: Purify cDNA and circularize using Circligase II.
- Linearization & PCR: Re-linearize cDNA and PCR amplify with Illumina-compatible primers.
- Sequencing: Perform high-depth sequencing on an Illumina platform.
Protocol 2: PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced CLIP)
Principle: Incorporation of 4-thiouridine (4SU) followed by crosslinking at 365 nm, inducing T-to-C transitions in sequenced cDNA that mark interaction sites.
Materials & Reagents:
- 4-thiouridine (4SU): Photoactivatable ribonucleoside.
- UV-A Crosslinker (365 nm)
- 5' RNA Adaptor (Phosphorylated)
- T4 RNA Ligase 1: For 5' adaptor ligation.
- α-[³²P]-ATP: For radioactive labeling of the 5' adaptor (optional for visualization).
- Urea-SDS Sample Buffer: For gel loading.
- TRIzol Reagent: For RNA extraction post-proteinase K.
- High-Fidelity PCR Enzyme
Procedure:
- 4SU Labeling: Incubate cells with 100-500 µM 4SU for one cell cycle (e.g., 16 hours).
- Crosslinking: Wash cells and irradiate with 365 nm UV-A (0.15-0.6 J/cm²).
- Lysis & RNase Digestion: Lyse in standard RIPA buffer and digest with RNase T1.
- Immunoprecipitation: Perform IP with specific antibody.
- 3' Dephosphorylation: Treat beads with T4 PNK (without ATP).
- 5' Adaptor Ligation: Radiolabel 5' adaptor with γ-[³²P]-ATP and PNK. Ligate to RNA using T4 RNA Ligase 1.
- Gel Purification: Run complex on SDS-PAGE, transfer to nitrocellulose, expose, and excise band.
- RNA Recovery: Digest with Proteinase K, extract RNA with TRIzol.
- Reverse Transcription & PCR: Use RT primer with partial Illumina adaptor. PCR amplify full library.
- Sequencing & Analysis: Sequence and identify T-to-C transitions in clusters to define binding sites.
Visualization of Workflows
Title: iCLIP Experimental Workflow
Title: PAR-CLIP Experimental Workflow
Title: CLIP Protocol Selection Decision Path
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CLIP-Seq Protocols
| Reagent | Function | Protocol Applicability |
|---|---|---|
| RNase I (or T1) | Creates RNA footprints by partial digestion; defines minimal binding region. | All (HITS-CLIP, PAR-CLIP, iCLIP) |
| Protein A/G Magnetic Beads | Solid support for antibody-mediated capture of RNP complexes. | All |
| T4 Polynucleotide Kinase (PNK) | 5' phosphorylation and 3' dephosphorylation of RNA for adaptor ligation. | All |
| Pre-Adenylated 3' Adaptor (L3-App) | Enabled by truncated T4 Rnl2, allows efficient ligation without ATP to prevent circularization. | iCLIP, modern HITS-CLIP |
| 4-Thiouridine (4SU) | Photoactivatable nucleoside precursor; incorporated into RNA for efficient 365 nm crosslinking. | PAR-CLIP exclusively |
| CircLigase II ssDNA Ligase | Catalyzes intramolecular ligation (circularization) of single-stranded DNA cDNAs. | iCLIP |
| Proteinase K | Digests the protein component of RNP complexes to release crosslinked RNA for downstream steps. | All |
| Nitrocellulose Membrane | Binds proteins irreversibly; used to purify RNP complexes away from free RNA/antibody after SDS-PAGE. | All |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR for final library amplification, minimizing bias and errors. | All (Library Prep) |
The study of RNA-binding proteins (RBPs) is central to understanding post-transcriptional gene regulation. The foundation of modern RBP-RNA interaction mapping was built by crosslinking and immunoprecipitation (CLIP) techniques, primarily HITS-CLIP, PAR-CLIP, and iCLIP. These protocols enabled genome-wide identification of RBP binding sites but were constrained by inefficiencies in RNA adapter ligation, high background, and limited quantitative accuracy. This evolution addresses these constraints, with emerging protocols like irCLIP and hiCLIP offering refined solutions for specific biological questions. This document details these novel methodologies, positioning them within the broader CLIP-seq thesis as specialized tools for enhanced resolution, efficiency, and application scope.
Comparative Analysis of CLIP Methodologies
The table below summarizes the core quantitative and methodological distinctions between established and emerging CLIP protocols.
Table 1: Comparative Analysis of Key CLIP-Seq Methodologies
| Feature | HITS-CLIP | PAR-CLIP | iCLIP | irCLIP | hiCLIP |
|---|---|---|---|---|---|
| Crosslink Type | UV-C (254 nm) | 4-Thiouridine + UV-A (365 nm) | UV-C (254 nm) | UV-C (254 nm) | UV-C (254 nm) |
| Key Innovation | High-throughput sequencing | T-to-C transitions mark sites | cDNA truncation at crosslink | Infrared dye-labeled adapters | Circularization-based ligation |
| Primary Advantage | Robust, widely adopted | Nucleotide-resolution mapping | Single-nucleotide resolution mapping | Dramatically reduced adapter dimer background | Efficient, single-step adapter ligation |
| Typical Efficiency (Adapter Ligation) | ~10-20% | ~10-20% | ~10-20% | >50% | >70% |
| Primary Application Niche | General RBP mapping | Precise binding site identification | Protein-RNA interaction footprinting | High-sensitivity, low-input, quantitative studies | Mapping of RNA-RNA duplexes (e.g., miRNA-target, IncRNA interactions) |
| Key Limitation | Low resolution, high background | Requires nucleotide analog incorporation | Complex workflow, lower yield | Requires IR scanner for gel excision | Specialized for proximal RNA pairs |
Detailed Application Notes and Protocols
irCLIP (Infrared-CLIP) Protocol
Principle: irCLIP replaces conventional radioactive or fluorescent labels with infrared dye (IR)-conjugated adapters. This allows precise gel excision using an infrared scanner, virtually eliminating contamination from adapter dimers, which are not IR-labeled and thus invisible in the IR channel.
Detailed Methodology:
- In Vivo Crosslinking & Lysis: UV-C crosslink cells (254 nm, 400 mJ/cm²). Lyse in stringent RIPA buffer.
- Partial RNA Digestion & Immunoprecipitation: Treat lysate with low-concentration RNase I (e.g., 0.05 U/µl) to generate RNA fragments. Perform IP with protein-specific antibody.
- 3’ Dephosphorylation & Ligation: Dephosphorylate RNA 3' ends with PNK. Ligate a pre-adenylated, IR800-labeled 3' adapter using T4 RnI2tr ligase.
- Radiolabeling & Separation: Label the 5' end with γ-³²P-ATP using PNK. Resolve RBP-RNA complexes on a Bis-Tris NuPAGE gel.
- IR-Based Excision: Transfer to a nitrocellulose membrane. Visualize the membrane using both a phosphorimager (for ³²P signal) and an IR scanner (Odyssey or similar). Overlay images; excise the protein-RNA complex band only where the ³²P signal overlaps with the IR signal. This excludes adapter-dimer contamination.
- Proteinase K Digestion & Purification: Elute and digest RNA with proteinase K. Purify RNA, reverse transcribe, and prepare library for sequencing.
Visualization: irCLIP Gel Excision Strategy
hiCLIP (Hybrid iCLIP) Protocol
Principle: hiCLIP is designed to capture RNA-RNA duplexes bound by an RBP (e.g., AGO, Staufen). It uses intra-molecular RNA circularization to ligate two RNA segments that are in close proximity due to the RBP-mediated duplex, enabling direct sequencing of the hybrid pair.
Detailed Methodology:
- Crosslinking, Digestion & IP: Perform standard UV-C crosslinking, mild RNase digestion, and immunoprecipitation.
- Proximity Ligation (Key Step): While RNA is still crosslinked to the protein on beads, use T4 RNA Ligase 1 to perform an intra-molecular ligation. This circularizes the RNA fragment, joining the two ends of the duplex region.
- Linearization & Adapter Ligation: Reverse crosslink and release RNA. Linearize the circular RNA by annealing an oligonucleotide complementary to the ligated junction and digesting with RNase H. This creates a unique sequence representing the duplex junction.
- Library Construction: Ligate standard sequencing adapters to the linearized fragment, amplify, and sequence. The junction sequence reveals the two interacting RNA segments.
Visualization: hiCLIP Proximity Ligation Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Advanced CLIP
| Reagent / Material | Function | Protocol Relevance |
|---|---|---|
| IRDye 800CW 3' Adapter | Infrared-labeled oligonucleotide for specific, low-background detection. | irCLIP: Enables clean gel excision by eliminating adapter dimer contamination. |
| T4 RNA Ligase 2, truncated (RnI2tr) | Catalyzes ligation of pre-adenylated 3' adapters to RNA 3'-OH. | irCLIP, iCLIP: Essential for efficient, specific 3' adapter ligation. |
| T4 RNA Ligase 1 | Catalyzes intra- and inter-molecular RNA ligation. | hiCLIP: Critical for proximity ligation of duplex ends to form circular RNA. |
| 4-Thiouridine (4sU) | Photosensitive nucleoside analog for incorporation into nascent RNA. | PAR-CLIP: Induces T-to-C transitions upon UV-A crosslinking for precise site mapping. |
| RNase I (Low Concentration) | Non-specific endoribonuclease for generating random RNA fragments of optimal size. | All CLIP variants: Creates uniformly sized RNA footprints for resolution. |
| Phos-tag Acrylamide Gels | Acrylamide bound to phosphate-binding tag for mobility shift assays. | Advanced Analysis: Can be used to monitor phosphorylation state of RBPs during CLIP optimization. |
| Magnetic Protein A/G Beads | Solid-phase support for antibody-based immunoprecipitation. | All CLIP variants: Enables efficient pull-down of RBP-RNA complexes. |
Conclusion
CLIP-seq technologies have revolutionized our understanding of the RNA-binding proteome and the regulatory codes embedded in RNA sequences. HITS-CLIP remains a robust, widely adopted standard, PAR-CLIP offers nucleotide-resolution through mutation signatures, and iCLIP provides superior mapping of crosslink sites. The choice of protocol is not one-size-fits-all but depends on the biological question, the RBP's characteristics, and practical laboratory considerations. Successful implementation hinges on meticulous optimization, rigorous controls, and complementary validation. Future directions point towards higher throughput (enhanced CLIP), single-cell applications, and integration with structural and functional assays to move from mapping interactions to deciphering mechanistic logic. For biomedical research, these refined maps of RBP-RNA interactions are essential for uncovering novel therapeutic targets in diseases driven by post-transcriptional dysregulation, such as cancer, neurodegeneration, and metabolic disorders.