RNA-seq vs qPCR: A Modern Guide to Differential Expression Validation
This article provides a comprehensive framework for researchers and drug development professionals navigating the complementary roles of RNA-seq and qPCR in differential expression analysis.
RNA-seq vs qPCR: A Modern Guide to Differential Expression Validation
Abstract
This article provides a comprehensive framework for researchers and drug development professionals navigating the complementary roles of RNA-seq and qPCR in differential expression analysis. It covers the foundational principles of each technology, explores their optimal applications from discovery to validation, and delivers practical troubleshooting strategies. By synthesizing evidence from large-scale benchmarking studies, it offers clear guidance on experimental design, data analysis pipelines, and the critical question of when orthogonal validation is necessary to ensure robust, reproducible results in biomedical research.
Understanding the Core Technologies: From qPCR Gold Standard to RNA-seq Discovery
In the field of gene expression analysis, the debate between adopting comprehensive RNA-sequencing (RNA-seq) technologies and established targeted methods remains active. While next-generation sequencing provides an unbiased, genome-wide view of the transcriptome, its accuracy for quantifying specific genes of interest requires rigorous validation. Within this context, quantitative PCR (qPCR) maintains its position as the established gold standard for targeted gene quantification, offering unparalleled accuracy, sensitivity, and reproducibility for validating gene expression data. This guide objectively compares the performance of qPCR against RNA-seq, presenting experimental data that underscores their respective strengths in research and drug development.
Methodological Foundations: How qPCR and RNA-seq Work
The fundamental differences in how qPCR and RNA-seq quantify gene expression underlie their performance characteristics. The workflows below illustrate the distinct steps involved in each process.
qPCR Workflow
RNA-seq Workflow
qPCR operates on the principle of amplifying a specific DNA target using sequence-specific primers, with fluorescence accumulation monitored in real-time. The cycle at which fluorescence crosses a threshold (Cq) is inversely proportional to the starting quantity of the target [1]. This direct relationship between signal and target concentration provides a highly precise quantification method.
RNA-seq utilizes high-throughput sequencing to capture fragments from the entire transcriptome. The resulting reads are mapped to a reference genome or transcriptome, and expression levels are inferred based on read counts [2]. This approach provides a comprehensive view but introduces mapping ambiguities and computational complexities that can affect quantification accuracy, especially for polymorphic gene families like HLA [3].
Performance Comparison: Quantitative Experimental Data
Direct comparisons between qPCR and RNA-seq reveal important differences in their quantification performance. The following table summarizes key findings from controlled studies that benchmarked these technologies head-to-head.
Table 1: Quantitative Comparison of qPCR vs. RNA-seq Performance
| Performance Metric | qPCR Performance | RNA-seq Performance | Experimental Context |
|---|---|---|---|
| Expression Correlation | Reference Standard | Moderate correlation (rho: 0.20-0.53) for HLA genes [3] | HLA class I gene expression in PBMCs from 96 healthy donors [3] |
| Fold Change Concordance | Reference Standard | 80-85% of genes show consistent fold changes with qPCR [2] | MAQCA/MAQCB reference samples; 18,080 protein-coding genes [2] |
| Non-concordant Genes | Reference Standard | 15-20% of genes show discordant differential expression [4] | Analysis of five RNA-seq workflows vs. qPCR [2] [4] |
| Technology-specific Biases | Minimal | Specific gene sets with inconsistent expression; typically shorter, lower expressed genes with fewer exons [2] | Systematic benchmarking using whole-transcriptome qPCR data [2] |
| Dynamic Range | High (6-8 orders of magnitude with proper validation) [5] | Broader in theory but limited for low-abundance transcripts | Dilution series with known standards [5] |
The moderate correlation between qPCR and RNA-seq for highly polymorphic HLA genes highlights the particular challenges RNA-seq faces with complex gene families [3]. While approximately 85% of genes show consistent fold-change relationships between the technologies, the remaining 15% discordance rate necessitates careful validation for key targets [2] [4].
Experimental Protocols for Method Validation
qPCR Assay Validation Protocol
For reliable qPCR results, the following validation steps must be implemented according to MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines [5] [6]:
- Inclusivity and Exclusivity Testing: Verify that primers detect all intended target variants (inclusivity) and do not amplify non-targets (exclusivity) through both in silico analysis and experimental testing [5].
- Dynamic Range and Efficiency: Prepare a seven-point 10-fold dilution series of the target template in triplicate. The assay should demonstrate a linear dynamic range of 6-8 orders of magnitude with amplification efficiencies between 90-110% [5].
- Limit of Detection (LOD) and Quantification (LOQ): Determine the lowest concentration that can be detected (LOD) and reliably quantified (LOQ) with acceptable precision [5].
- Precision and Accuracy: Assess intra- and inter-assay variability, with accuracy measured as closeness to the true value and precision as agreement between replicates [6].
RNA-seq Validation Using qPCR
When using qPCR to validate RNA-seq findings:
- Reference Gene Selection: Identify stable, highly expressed reference genes specifically for your experimental system. RNA-seq data itself can be mined to identify optimal reference genes, moving beyond traditional housekeeping genes that may vary under different conditions [7] [8].
- Candidate Gene Prioritization: Focus validation efforts on genes with fold changes >2 and adequate expression levels (e.g., log2 TPM >5) to ensure reliable detection by qPCR [8].
- Sample Overlap: Ideally, use the same RNA samples for both RNA-seq and qPCR validation to remove biological variation from the comparison [3].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential Reagents for qPCR Experiments
| Reagent/Solution | Function | Critical Considerations |
|---|---|---|
| Sequence-Specific Primers | Amplify target sequence | Must be validated for inclusivity/exclusivity; designed to avoid secondary structures [5] |
| Fluorescent Detection System | (e.g., SYBR Green, hydrolysis probes) | Enable real-time monitoring of amplification; probes offer higher specificity [1] |
| Reverse Transcriptase Enzyme | Convert RNA to cDNA | Efficiency impacts overall quantification accuracy; must be consistent across samples [6] |
| Quantification Standards | (e.g., synthetic oligos, purified amplicons) | Create standard curve for absolute quantification; should mimic sample amplification [1] |
| RNA Isolation Reagents | Purify intact RNA from samples | Quality critical; must remove genomic DNA contamination [6] |
| Reference Genes | Normalize technical and biological variation | Must be stably expressed across experimental conditions; not necessarily traditional housekeepers [7] [8] |
Decision Framework: When to Use Each Technology
The relationship between qPCR and RNA-seq is often complementary rather than competitive. The following diagram illustrates their interplay in a rigorous gene expression study.
Appropriate Applications for qPCR:
- Targeted validation of genes identified in discovery-phase RNA-seq studies
- High-throughput clinical screening of established biomarker panels
- Regulated environments requiring validated assays (e.g., diagnostic test development)
- Studies with limited sample material where maximum sensitivity is required
- Rapid turnaround projects with constrained computational resources
Appropriate Applications for RNA-seq:
- Discovery-phase research to identify novel transcripts and splice variants
- Comprehensive transcriptome profiling without prior knowledge of targets
- Studies of structural variants, fusion genes, or allele-specific expression
- Organisms without established genomes (using de novo assembly approaches)
In the evolving landscape of gene expression analysis, qPCR maintains its critical role as the gold standard for targeted quantification. Its superior accuracy, sensitivity, and reproducibility make it indispensable for validating RNA-seq findings, particularly for clinically significant targets. While RNA-seq provides an unparalleled discovery platform, the 15-20% discordance rate between the technologies necessitates orthogonal validation for key results. Researchers should view these technologies as complementary components of a rigorous gene expression workflow, leveraging the strengths of each to generate reliable, reproducible data that advances scientific understanding and drug development.
For decades, gene expression analysis was constrained by targeted approaches, with quantitative real-time PCR (qRT-PCR) serving as the gold standard for measuring the expression of a limited number of pre-selected genes. While qRT-PCR offers excellent sensitivity and reproducibility for focused studies, its reliance on a priori knowledge of target genes inherently biases discovery and prevents a holistic understanding of cellular states [9]. The advent of RNA sequencing (RNA-seq) has fundamentally transformed this paradigm by providing an unbiased, genome-wide platform for transcriptome exploration. This technology enables researchers to quantify gene expression across the entire transcriptome, detect novel transcripts, identify alternative splicing events, and discover fusion genes—all without any prior assumptions about the genome [10].
This guide objectively compares the performance of RNA-seq against established technologies like qRT-PCR and microarrays, providing supporting experimental data and detailed methodologies to help researchers, scientists, and drug development professionals navigate this powerful landscape.
Technology Comparison: RNA-seq vs. qPCR and Microarrays
Key Characteristics at a Glance
The table below summarizes the core differences between RNA-seq and its primary alternatives.
Table 1: Comparison of Key Gene Expression Analysis Technologies
| Feature | RNA-seq | qPCR | Microarrays |
|---|---|---|---|
| Throughput | High (entire transcriptome) | Medium (tens to hundreds of targets) | High (known transcriptome) |
| Prior Knowledge Required | No (can discover novel features) | Yes (specific primers/probes needed) | Yes (probes designed from known sequences) |
| Dynamic Range | >9,000-fold [10] | ~7-log range [9] | ~3,000-fold |
| Sensitivity | High (can detect low-abundance transcripts) | Very High (can detect single copies) | Lower (background noise limitations) |
| Applications | Differential expression, novel transcripts, splicing, fusions, allele-specific expression [10] | Targeted differential expression, validation [9] | Differential expression (known transcripts) |
| Quantitative Nature | Digital (read counting) | Analog (fluorescence-based) | Analog (fluorescence-based) |
| Cost per Sample | Higher | Lower for limited targets | Moderate |
The Complementary Role of qPCR in RNA-seq Workflows
Rather than being a simple replacement, qPCR often works in tandem with RNA-seq to generate trustworthy results [9]. Its role is critical both upstream and downstream of an RNA-seq experiment:
- Upstream: TaqMan qPCR is commonly used to check cDNA integrity prior to library preparation for NGS, ensuring that input material is of sufficient quality [9].
- Downstream: qPCR remains the go-to method for validating RNA-seq results. For follow-up studies focusing on a targeted panel of transcripts discovered during the NGS screen, qPCR is the gold-standard technology [9] [8].
RNA-seq Experimental Design and Data Analysis
Core Workflow and Essential Bioinformatics Tools
A standard RNA-seq analysis involves several sequential steps, with critical decisions required at each stage. The diagram below illustrates this workflow and the common tool choices.
Figure 1: The core stages of an RNA-seq data analysis workflow and the associated bioinformatics tools for each step [11] [10].
Validating Differential Expression Analysis Methods
Given the variety of statistical tools available for identifying differentially expressed genes (DEGs), independent validation is crucial. One study experimentally validated DEGs identified by four common methods (Cuffdiff2, edgeR, DESeq2, and TSPM) using high-throughput qPCR on independent biological samples [12].
Table 2: Performance of DEG Analysis Methods Validated by qPCR
| Method | Sensitivity | Specificity | False Positivity Rate | False Negativity Rate | Positive Predictive Value |
|---|---|---|---|---|---|
| edgeR | 76.67% | 90.91% | 9% | 23.33% | 90.20% |
| Cuffdiff2 | 51.67% | 45.45% | High (54.55%) | 48.33% | 39.24% |
| DESeq2 | 1.67% | 100% | 0% | 98.33% | 100% |
| TSPM | 5.00% | 90.91% | 9% | 95% | 37.50% |
The results highlight a significant trade-off: DESeq2 was the most specific but least sensitive method, while Cuffdiff2 generated a high false positivity rate. Among the tested methods, edgeR demonstrated the best balance of sensitivity and specificity, with a high positive predictive value, making its findings most likely to be confirmed by an independent gold-standard method like qPCR [12]. This underscores the need for careful tool selection based on the research goals—whether prioritizing novel discovery (favoring sensitivity) or confident validation of a smaller gene set (favoring specificity).
The Validation Loop: Integrating RNA-seq and qPCR
The relationship between RNA-seq and qPCR is not competitive but collaborative. The following diagram outlines a robust workflow for using these technologies together to ensure discovery and validation.
Figure 2: An integrated RNA-seq and qPCR workflow for discovery and validation, highlighting the critical step of appropriate gene selection [9] [8].
A critical, often neglected step in this process is the informed selection of reference genes for qPCR validation. Traditional housekeeping genes (e.g., ACTB, GAPDH) may exhibit variable expression under different biological conditions, leading to normalization errors and misinterpretation of results [8]. Tools like the Gene Selector for Validation (GSV) software now leverage RNA-seq data itself to identify the most stable, highly expressed reference genes and the most variable candidate genes for validation, ensuring reliable and cost-effective qPCR experiments [8].
The Scientist's Toolkit: Essential Reagents and Platforms
Table 3: Key Research Reagent Solutions and Platforms for RNA-seq
| Item / Platform | Function / Application | Key Considerations |
|---|---|---|
| Library Prep Kits (e.g., NuGEN Ovation) | Convert RNA into sequence-ready cDNA libraries. | Protocol efficiency, bias correction, compatibility with low-input RNA. |
| TaqMan qPCR Assays | Validate RNA-seq results and check cDNA integrity. | Predesigned assays for most exon-exon junctions; require variant-specific design for isoform detection [9]. |
| Alignment & Quantification Tools (STAR, HISAT2, Salmon) | Map reads to a reference and quantify gene/transcript abundance. | STAR is fast but memory-intensive; HISAT2 has a smaller footprint; Salmon is alignment-free and fast [11]. |
| Differential Expression Software (DESeq2, edgeR, Limma-voom) | Statistically identify genes changed between conditions. | DESeq2 good for small-n studies; edgeR for well-replicated experiments; Limma-voom excels with large cohorts [11] [12]. |
| Integrated Commercial Platforms (Partek Flow, CLC Genomics) | GUI-based, end-to-end analysis from raw data to results. | Reduce bioinformatics burden; offer validated workflows for regulated environments [11]. |
| Single-Cell Platforms (Nygen, BBrowserX) | Analyze transcriptomes at single-cell resolution. | Handle cell clustering, annotation, and multi-omics integration; often cloud-based with AI-powered insights [13]. |
RNA-seq has firmly established itself as the premier technology for unbiased, genome-wide transcriptome exploration, enabling discoveries that are simply impossible with targeted approaches. Its power, however, does not render older methods like qPCR obsolete. Instead, a synergistic workflow, where RNA-seq drives hypothesis-free discovery and qPCR provides robust, targeted validation, represents the current gold standard in gene expression research. As the field continues to evolve with lower costs, longer reads, and integrated single-cell and spatial modalities [13], this foundational principle of collaborative technology application will continue to ensure the generation of reliable and impactful biological insights.
In transcriptomics research, a central question has long been whether gene expression data obtained through high-throughput sequencing technologies require validation by targeted amplification methods like quantitative PCR (qPCR). Next-generation sequencing (NGS), particularly RNA sequencing (RNA-seq), provides an unbiased, genome-wide view of the transcriptome, while qPCR offers high sensitivity and specificity for quantifying a limited number of targets. This guide objectively compares the technical performance of these two paradigms—broad sequencing and targeted amplification—within the context of differential gene expression analysis, providing researchers with the data needed to inform their validation strategies.
Core Technological Principles
Amplification-Based Quantification: qPCR
Quantitative PCR (qPCR) is a targeted method for gene expression analysis that relies on the enzymatic amplification of specific cDNA sequences. Its function is to quantify the abundance of a transcript by measuring the amplification kinetics, with the cycle threshold (Cq) indicating the starting quantity. The process involves reverse transcribing RNA into cDNA, followed by thermal cycling that uses DNA polymerase to exponentially amplify target sequences, with fluorescence intensity measured in real time to track product accumulation [4] [8].
Sequencing-Based Quantification: RNA-Seq
RNA sequencing (RNA-seq) is a comprehensive method that determines the sequence of nucleotides in a population of RNA molecules. Its primary function is to identify and quantify the multitude of RNA transcripts in a sample, from known genes to novel isoforms. In the predominant short-read sequencing approach (e.g., Illumina), the workflow involves fragmenting RNA, converting it to cDNA, attaching adapters, and then using sequencing-by-synthesis chemistry on a massively parallel scale to generate billions of short reads that are subsequently mapped to a reference genome for quantification [14] [15] [16].
Direct Performance Comparison in Differential Expression
Independent benchmarking studies have systematically compared the performance of RNA-seq and qPCR for identifying differentially expressed genes, providing critical data for evaluating the need for validation.
A comprehensive benchmark study compared five RNA-seq analysis workflows against whole-transcriptome qPCR data for over 18,000 protein-coding genes. The results demonstrated a high overall correlation for gene expression fold changes between RNA-seq and qPCR, with Pearson correlation coefficients (R²) ranging from 0.927 to 0.934 across different computational methods [2].
The study revealed that approximately 85% of genes showed consistent differential expression calls between RNA-seq and qPCR. However, about 15% of genes showed non-concordant results, where the two methods disagreed on differential expression status or direction. Importantly, of these non-concordant genes, 93% had fold-change differences (ΔFC) of less than 2, and approximately 80% had ΔFC less than 1.5, indicating that most discrepancies were of small magnitude [4] [2].
Characteristics of Problematic Genes
Research indicates that the small fraction of genes with severe discrepancies (approximately 1.8%) are typically characterized by specific features. These non-concordant genes with ΔFC > 2 were predominantly shorter, lower expressed genes with fewer exons compared to genes with consistent expression measurements [4] [2].
Figure 1: Concordance and discrepancy patterns between RNA-seq and qPCR in differential expression analysis.
Experimental Design and Protocol Considerations
RNA-Seq Experimental Workflows
Multiple RNA-seq library preparation and sequencing protocols exist, each with distinct technical considerations that impact their performance relative to qPCR.
Short-Read vs. Long-Read Sequencing
The recent SG-NEx project systematically benchmarked five RNA-seq protocols, including short-read cDNA sequencing, Nanopore long-read direct RNA, amplification-free direct cDNA, PCR-amplified cDNA sequencing, and PacBio IsoSeq. The study found that while short-read data generate robust estimates for gene expression, long-read sequencing more reliably identifies major isoforms and complex transcriptional events, though with different cost and throughput considerations [17].
Amplification in Library Preparation
A rigorous 2024 comparison of Nanopore direct cDNA and PCR-cDNA sequencing for bacterial transcriptomes demonstrated that PCR-based amplification substantially improves sequencing yield with largely unbiased assessment of core gene expression. However, a small risk of technical bias was identified, which appeared greater for genes with unusually high (>52%) or low (<44%) GC content [18].
qPCR Validation Protocols
For rigorous validation of RNA-seq results by qPCR, specific methodological considerations are essential.
Reference Gene Selection
Traditional use of housekeeping genes (e.g., actin, GAPDH) as reference genes for normalization is problematic, as their expression can vary across biological conditions. Computational tools like Gene Selector for Validation (GSV) have been developed to identify optimal reference genes directly from RNA-seq data based on stability and expression level across experimental conditions [8].
The GSV algorithm applies multiple filtering criteria to select optimal reference genes:
- Expression >0 TPM in all samples
- Standard variation of log2(TPM) <1
- No exceptional expression in any library (<2× average log2 expression)
- Average log2 expression >5
- Coefficient of variation <0.2 [8]
Technical Comparison Tables
Performance Characteristics for Differential Expression
Table 1: Comparative performance of RNA-seq and qPCR for differential expression analysis
| Performance Metric | RNA-Seq | qPCR | Experimental Evidence |
|---|---|---|---|
| Throughput | Genome-wide (all transcripts) | Targeted (dozens to hundreds) | [4] [2] |
| Fold Change Correlation | R² = 0.93-0.94 vs qPCR | Reference method | [2] |
| Concordance Rate | ~85% with qPCR | ~85% with RNA-seq | [4] [2] |
| Problematic Genes | Shorter, lower expressed genes with fewer exons | Less affected by transcript features | [4] [2] |
| Dynamic Range | Broad (~5-6 orders of magnitude) | Very broad (>7 orders of magnitude) | [4] |
Technical Specifications and Methodological Considerations
Table 2: Technical specifications and methodological requirements
| Characteristic | RNA-Seq | qPCR | Notes |
|---|---|---|---|
| Sample Input | 10 ng - 1 μg total RNA | 1 pg - 100 ng total RNA | [17] [18] |
| Amplification Required | Yes (library preparation) | Yes (target amplification) | [15] [18] |
| Multiplexing Capacity | Very high (multiple samples per run) | Moderate (dozens of targets per run) | [15] |
| Hands-on Time | Moderate to high | Low to moderate | [18] |
| Data Analysis Complexity | High (bioinformatics expertise) | Low to moderate (standard curves) | [15] [8] |
| Cost per Sample | $50 - $1000+ | $5 - $50 | Platform dependent |
Decision Framework for Validation Requirements
When qPCR Validation Is Recommended
Evidence suggests that qPCR validation provides the most value in specific scenarios:
- When an entire biological conclusion rests on differential expression of only a few genes
- For genes with low expression levels and/or small fold changes (<1.5-2)
- When extending findings to additional samples, strains, or conditions beyond the original RNA-seq experiment [4]
- For specific challenging gene families, such as HLA genes, where moderate correlations (rho = 0.2-0.53) have been observed between RNA-seq and qPCR [3]
When RNA-Seq Stands Alone
Under optimal experimental conditions, RNA-seq data may not require qPCR validation:
- When studies include a sufficient number of biological replicates
- When following established minimum information guidelines (e.g., MINSEQE for sequencing, MIABiE for biofilm experiments)
- When using state-of-the-art data analysis pipelines
- For genes with moderate to high expression levels and substantial fold changes [4]
Figure 2: Decision framework for determining when qPCR validation of RNA-seq results is most beneficial.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagents and their applications in amplification and sequencing workflows
| Reagent/Solution | Function | Application Context |
|---|---|---|
| Poly(A) Polymerase | Adds poly(A) tails to bacterial mRNA | Nanopore sequencing of prokaryotic transcriptomes [18] |
| RNase Inhibitor | Prevents RNA degradation during library prep | All RNA-seq workflows, especially long protocols [18] |
| Oligo(dT) Primers | Binds to poly(A) tails for cDNA synthesis | mRNA enrichment in eukaryotic transcriptomics [18] |
| rRNA Depletion Kits | Removes abundant ribosomal RNA | Bacterial RNA-seq to increase mRNA sequencing depth [18] |
| Reverse Transcriptase | Synthesizes cDNA from RNA templates | Essential first step in both qPCR and RNA-seq [4] [8] |
| DNA Polymerase | Amplifies DNA templates | qPCR and amplification-based sequencing libraries [18] |
| Unique Molecular Identifiers | Tags individual molecules | Multiplexing samples in NGS library preparation [15] |
The choice between amplification and sequencing technologies for gene expression analysis involves balancing throughput, precision, and practical considerations. RNA-seq has matured to provide highly reliable differential expression results for the majority of genes, potentially reducing the need for systematic qPCR validation. However, targeted amplification remains indispensable for validating critical findings, particularly for low-expressed genes or those with challenging sequence features. By understanding the specific technical differences and performance characteristics outlined in this guide, researchers can make evidence-based decisions about when amplification-based validation is truly necessary, optimizing their experimental workflows for robust and efficient transcriptome analysis.
In differential expression research, the choice between quantitative polymerase chain reaction (qPCR) and RNA sequencing (RNA-Seq) is foundational. While RNA-Seq provides a comprehensive, hypothesis-free view of the transcriptome, qPCR offers a sensitive, targeted, and highly precise approach for quantifying specific transcripts. The prevailing practice of using qPCR to validate RNA-Seq results is common, yet the relationship between these technologies is more nuanced than simple verification. A deeper understanding of their respective strengths, limitations, and optimal application spaces enables researchers to design more efficient and cost-effective studies. This guide objectively compares their performance based on experimental data, detailing methodologies to inform strategic decisions in biomedical research and drug development.
Performance and Capability Comparison
The following tables summarize the core technical and operational characteristics of qPCR and RNA-Seq, providing a direct comparison of their performance.
Table 1: Key Technical and Performance Specifications
| Feature | qPCR | Bulk RNA-Seq | Single-Cell/Nucleus RNA-Seq (sc/snRNA-Seq) |
|---|---|---|---|
| Throughput | Low to medium (tens to hundreds of targets) | High (entire transcriptome) | Very High (thousands to millions of cells) |
| Sensitivity | Very High (can detect single copies) [19] | High (requires ~20-30M reads/sample for robust DGE) [20] | Low at single-cell level; improves with cell count [21] |
| Dynamic Range | ~7-8 logs | >5 logs | Constrained by high dropout rates [21] |
| Accuracy & Precision | High, mature technology | High for gene-level expression; lower for isoforms | Generally low at single-cell level [21] |
| Primary Application | Targeted quantification, validation | Discovery, differential expression, splicing | Cellular heterogeneity, rare cell types [21] [22] |
| Best Suited For | - Validating a limited number of genes- Low-abundance transcripts- High sample throughput studies | - Unbiased transcriptome discovery- Detecting novel transcripts/isoforms- Splice variant analysis | - Deconstructing cellular heterogeneity- Identifying novel cell types/states- Developmental trajectories |
Table 2: Cost and Workflow Considerations
| Consideration | qPCR | RNA-Seq (using Illumina TruSeq on NovaSeq S4 flow cell) |
|---|---|---|
| Cost per Sample (Library Prep & Sequencing) | Low (cost-effective for few targets) | ~$36.9 - $113.9, highly dependent on multiplexing and read depth [23] |
| Hands-on Time | Low (workflow is simple and fast) | ~3-4 days [23] |
| Data Analysis Complexity | Low (straightforward ΔΔCq method) | High (requires bioinformatics expertise) [20] [22] |
| Required Replicates | 3+ (standard for statistical power) | Minimum 3+; more needed for high variability [20] |
| Key Cost/Design Drivers | - Number of targets- Number of samples | - Library prep method ($24-$68.7/sample) [23]- Sequencing depth (5M-30M+ reads/sample) [23]- Level of multiplexing [23] |
Experimental Data and Validation Paradigms
When is qPCR Validation Appropriate?
The requirement for qPCR validation of RNA-Seq data is context-dependent. It is most appropriate in two key scenarios:
- For Independent Confirmation: When a second, orthogonal method is required to confirm a critical finding, such as for publication in a high-impact journal where reviewers demand confirmation via a different technical approach [24].
- For Underpowered RNA-Seq Studies: When the original RNA-Seq data is based on a small number of biological replicates, limiting the statistical power. Using qPCR to assay more samples for a focused set of targets can validate and extend the study findings [24].
A Case Study in Ovarian Cancer Diagnostics
A 2025 study on ovarian cancer detection provides a clear example of the complementary strengths of these technologies. Researchers used RNA-Seq as a discovery tool to analyze platelet RNA from patient blood samples, identifying a panel of 10 splice-junction-based biomarkers that differentiated ovarian cancer from benign conditions [25].
Subsequently, they developed a qPCR-based algorithm for clinical application. This targeted approach demonstrated 94.1% sensitivity and 94.4% specificity (AUC = 0.933) [25]. The study highlights a powerful workflow: using RNA-Seq's broad profiling capability for biomarker discovery, then leveraging qPCR's accessibility, low cost, and simplicity for a robust, deployable diagnostic test, especially where NGS is too costly for widespread use [25].
Detailed Experimental Protocols
Protocol: High-Sensitivity qPCR for Low-Abundance Transcripts (STALARD Method)
Background: Conventional RT-qPCR becomes unreliable for quantification cycle (Cq) values above 30-35 [19]. The STALARD (Selective Target Amplification for Low-Abundance RNA Detection) method overcomes this by incorporating a targeted pre-amplification step [19].
Methodology:
Primer Design:
- Design a Gene-Specific Primer (GSP) that matches the known 5'-end sequence of the target RNA (with T substituted for U). The GSP should have a Tm of ~62°C and 40-60% GC content.
- Synthesize a GSP-tailed oligo(dT) primer (GSoligo(dT)), which is an oligo(dT)24VN primer with the GSP sequence at its 5' end [19].
Reverse Transcription:
- Perform first-strand cDNA synthesis using 1 µg of total RNA and the GSoligo(dT) primer. This produces cDNA molecules that have the GSP sequence incorporated at both ends [19].
Targeted Pre-amplification:
- Perform a limited-cycle PCR (9-18 cycles) using only the GSP. This selectively amplifies the full-length cDNA of the target transcript.
- Reaction Setup:
- 1 µL of cDNA from step 2.
- 1 µL of 10 µM GSP.
- SeqAmp DNA Polymerase (Takara) [19].
- Thermal Cycling:
- 95°C for 1 min (initial denaturation).
- 9-18 cycles of: 98°C for 10 s, 62°C for 30 s, 68°C for 1 min/kb.
- 72°C for 10 min (final extension) [19].
Purification and Quantification:
- Purify the PCR product using AMPure XP beads.
- Use the purified product as the template for standard qPCR quantification with isoform-specific primers [19].
Protocol: Bulk RNA-Seq Library Preparation and Sequencing
Background: This protocol outlines the standard workflow for bulk RNA-Seq, which is the foundation for differential expression analysis [20].
Methodology:
RNA Extraction and QC:
- Extract total RNA using a solvent-based method (e.g., TRIzol, ~$2.2/sample) or a silica-based column kit (e.g., QIAgen RNeasy, ~$7.1/sample) [23].
- Assess RNA quality and integrity using an instrument like the Bioanalyzer. An RNA Integrity Number (RIN) ≥ 7 or distinct ribosomal RNA peaks are typically required [25].
Library Preparation:
- Option 1 (Standard): Use a kit like Illumina's TruSeq Stranded mRNA (~$64.4/sample) which involves mRNA enrichment, fragmentation, reverse transcription, adapter ligation, and PCR amplification [23].
- Option 2 (Cost-Effective): Use a 3'-end multiplexing kit like the Alithea MERCURIUS BRB-seq kit (~$24/sample), which uses early barcoding and pooling to drastically reduce costs [23].
- Critical Parameter: The number of PCR cycles during amplification must be optimized. Using more cycles than recommended for a given RNA input (e.g., <125 ng) leads to a high rate of PCR duplicates, reduced library complexity, and increased noise [26].
- Check final library quality and fragment size using a Bioanalyzer DNA chip [23].
Sequencing:
Technology Workflow Visualization
The following diagram illustrates the key procedural steps for RNA-Seq and qPCR, highlighting their divergent paths from sample to answer.
Diagram 1: Technology Workflow Comparison. This graph contrasts the comprehensive, sequencing-driven RNA-Seq pathway with the streamlined, amplification-focused qPCR pathway, showing their divergence based on the experimental goal.
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Reagents and Kits for RNA Expression Analysis
| Reagent/Kits | Primary Function | Example Products & Cost |
|---|---|---|
| RNA Extraction Kits | Isolate high-quality total RNA from samples. | - TRIzol (~$2.2/sample) [23]- QIAgen RNeasy Kit (~$7.1/sample) [23] |
| RNA Quality Control | Assess RNA integrity (RIN) and quantity. | - Agilent Bioanalyzer RNA-6000 Nano Kit (~$4.1/sample) [23] |
| qPCR Master Mix | Provides enzymes, dNTPs, and buffer for efficient and specific amplification. | - Not specified in search results, but numerous commercial options exist (e.g., from Bio-Rad, Thermo Fisher). |
| RNA-Seq Library Prep Kits | Convert RNA into sequencer-ready DNA libraries. | - Illumina TruSeq mRNA Stranded (~$64.4/sample) [23]- NEBnext Ultra II RNA (~$37/sample) [23]- Alithea MERCURIUS BRB-seq (~$19.7/sample) [23] |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences added to RNA fragments to tag and identify PCR duplicates [26]. | - Incorporated into many modern library prep kits. |
The decision between qPCR and RNA-Seq is not a matter of superiority but of strategic alignment with research goals. RNA-Seq is the undisputed tool for unbiased discovery, profiling entire transcriptomes, and detecting novel features. qPCR excels in targeted quantification, offering high sensitivity, low cost, and operational simplicity for validating key findings or conducting high-throughput screens of known targets.
A modern, robust approach involves using these technologies in concert: leveraging RNA-Seq's power for initial discovery and then employing qPCR's precision for validation and expansion on larger sample cohorts. Furthermore, best practices in experimental design—such as including sufficient biological replicates, optimizing PCR cycles to minimize duplicates, and utilizing UMIs—are critical for ensuring the reliability of data from either technology, ultimately leading to more reproducible and impactful scientific outcomes.
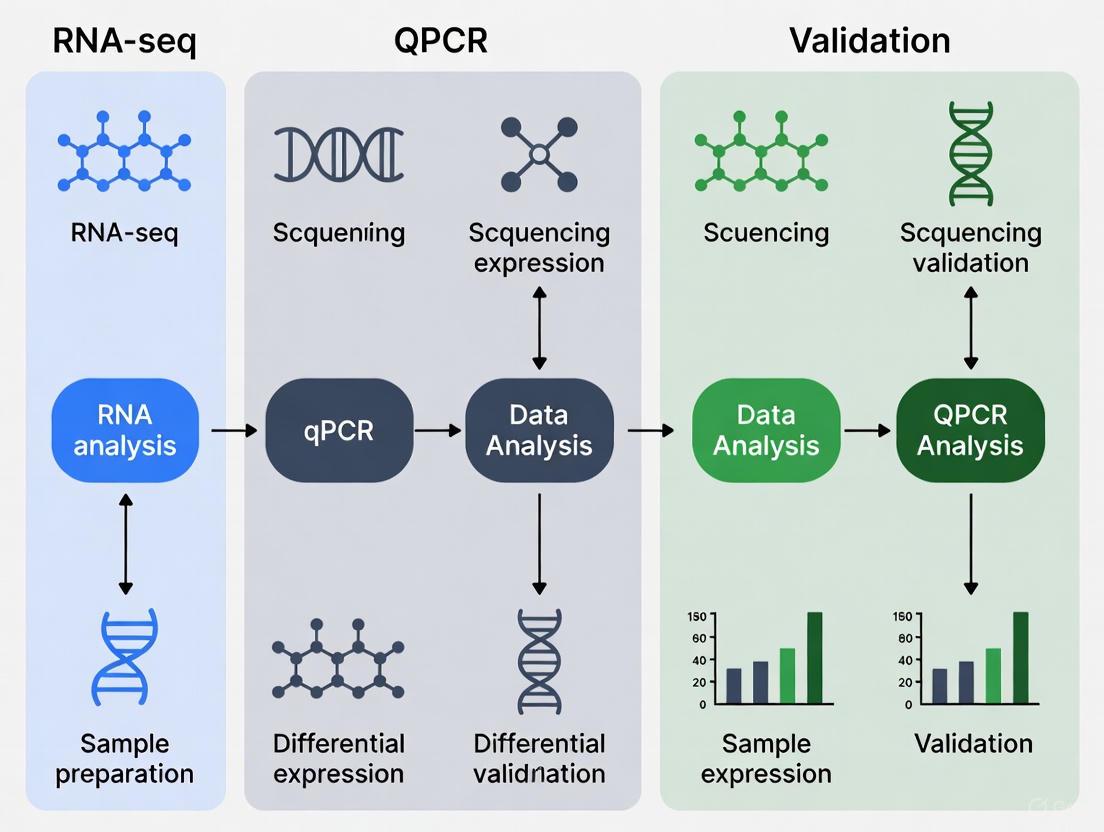
Designing Your Workflow: From RNA-seq Discovery to qPCR Confirmation
The debate in transcriptomics research often positions quantitative PCR (qPCR) and RNA sequencing (RNA-seq) as competing technologies. However, a more powerful approach emerges when they are used as complementary tools within an integrated workflow. RNA-seq provides an unbiased, genome-wide view of the transcriptome, enabling novel discovery, while qPCR delivers highly sensitive, specific, and reproducible quantification for targeted gene analysis. This guide objectively compares their performance and demonstrates how their strategic integration—using qPCR both upstream to ensure input quality and downstream to verify key findings—creates a robust framework for reliable gene expression research, ultimately strengthening experimental conclusions for drug development and clinical applications [9] [24].
Core Principles and Comparative Strengths
qPCR is a targeted technique that quantifies the amplification of specific cDNA sequences in real-time using fluorescent reporters. Its maturity, simplicity, and low operational cost make it the gold standard for validating a limited number of genes with high sensitivity and a wide dynamic range [9] [24].
RNA-seq is a discovery-oriented technology that involves converting RNA into a library of cDNA fragments, sequencing them using high-throughput platforms (e.g., Illumina, Nanopore, PacBio), and aligning the resulting millions of short reads to a reference genome to determine transcript abundance and structure [27] [28].
Table: Core Technical Comparison of qPCR and RNA-seq
| Feature | qPCR | RNA-seq |
|---|---|---|
| Throughput | Low (dozens to hundreds of targets) | High (entire transcriptome) |
| Target Selection | Requires a priori knowledge | Unbiased, capable of novel discovery |
| Sensitivity & Dynamic Range | High | Sufficient for most applications, though highly sensitive detection requires deep sequencing [9] |
| Primary Output | Cycle threshold (Cq) | Read counts (e.g., raw counts, TPM) |
| Key Advantage | High reproducibility, low cost per target, simple workflow | Comprehensive coverage, can detect novel transcripts, isoforms, and fusions [9] [28] |
| Typical Cost & Time | Lower cost and faster for studies with few targets/samples [9] | Higher cost and longer turnaround, especially when outsourced [9] |
The Integrated Workflow Diagram
The following diagram illustrates the synergistic workflow of using qPCR at critical points before and after RNA-seq.
Diagram: Integrated qPCR and RNA-seq Workflow. qPCR is used upstream to check cDNA quality before RNA-seq and downstream to validate key findings on new samples.
The Upstream Role: Using qPCR for Quality Control
The sensitivity and accuracy of RNA-seq are fundamentally dependent on the quality and quantity of the input RNA and cDNA [29]. Using qPCR upstream provides a functional quality check that is more specific than spectrophotometry.
Experimental Protocol: cDNA Integrity Check
This protocol is used prior to costly RNA-seq library preparation to confirm that reverse transcription has been successful.
- Step 1: RNA Quantification. Isolate total RNA using a dedicated kit (e.g., PureLink RNA Mini Kit, MagMAX-96 Total RNA Isolation Kit). Quantify RNA precisely using a fluorometric method like the Qubit RNA assay, which is more specific and sensitive for RNA than UV absorbance readings from a NanoDrop, as it minimizes interference from contaminants [29].
- Step 2: Reverse Transcription. Synthesize first-strand cDNA using a high-performance reverse transcriptase master mix (e.g., SuperScript IV VILO Master Mix). This step is critical for reducing amplification bias and ensuring superior linearity across a broad range of input material [29].
- Step 3: qPCR Quality Control. Perform qPCR on the resulting cDNA using TaqMan Gene Expression Assays targeting a set of stable, well-characterized housekeeping genes (e.g., GAPDH, ACTB). The criteria for passing this QC step include [9]:
- Low Cq Values: The quantification cycle (Cq) for control genes should be low and consistent across samples, indicating efficient cDNA synthesis from high-quality RNA.
- Minimal Replicate Variation: Technical replicates should show low variability (e.g., Cq standard deviation < 0.2).
- Step 4: Proceed to Library Prep. Only samples passing the cDNA QC threshold should proceed to RNA-seq library preparation, ensuring that sequencing resources are not wasted on degraded or inefficiently converted samples.
The Downstream Role: Using qPCR for Validation and Follow-up
Following RNA-seq and bioinformatic analysis, qPCR is deployed downstream to verify the expression patterns of a subset of critical genes, thereby bolstering confidence in the RNA-seq results.
Experimental Protocol: Validating Differential Expression
This protocol is for independently confirming the differential expression of key genes identified by RNA-seq.
- Step 1: Select Key Targets. From the RNA-seq results, select genes of high biological interest (e.g., most significantly differentially expressed genes, genes central to a key pathway). The number of targets is typically limited to what fits on a 96- or 384-well qPCR plate.
- Step 2: Choose an Independent Sample Set. For the most robust validation, perform qPCR on a new, larger set of biological replicates that were not used in the initial RNA-seq experiment. This validates both the technology and the underlying biology [24].
- Step 3: Perform qPCR with Rigorous Design. Use a pre-designed platform like TaqMan Array plates or cards for efficiency. The experiment must be designed with proper biological replication (e.g., n ≥ 3 per group) and technical replication (e.g., duplicates or triplicates per sample). It is crucial to use stable reference genes selected specifically for the biological system under study [8].
- Step 4: Data Analysis and Concordance Check. Analyze qPCR data using the ΔΔCq method. Determine concordance by comparing the direction (up/down-regulated) and magnitude (fold-change) of expression differences between the RNA-seq and qPCR results.
Evidence for Concordance and Its Limits
A comprehensive benchmark study analyzing over 18,000 protein-coding genes found a high level of concordance between RNA-seq and qPCR, with only about 1.8% of genes showing severe non-concordance. Notably, the majority of non-concordant results occurred in genes with low expression levels (fold-change < 2) [4]. This evidence supports the practice of using qPCR for validation, while also highlighting that validation is most critical when a study's conclusions hinge on a few genes, particularly those with low expression or small fold-changes [4] [24].
Table: Scenarios for Downstream qPCR Validation
| Scenario | Appropriate for qPCR Validation? | Rationale |
|---|---|---|
| Small number of RNA-seq replicates | Yes, highly appropriate [24] | qPCR on a larger sample set statistically confirms the biological effect. |
| Conclusions rely on a few key, low-expression genes | Yes, highly appropriate [4] | Confirms findings in a domain where RNA-seq pipelines can be variable [30]. |
| RNA-seq is a hypothesis-generating screen | Often unnecessary [24] | Resources are better directed toward functional protein-level studies. |
| Planning a larger, confirmatory RNA-seq study | Unnecessary [24] | The subsequent RNA-seq study itself serves as validation. |
Essential Research Reagent Solutions
The following table details key materials and their functions for executing the integrated workflow.
Table: Essential Reagents for the qPCR and RNA-seq Workflow
| Product Category | Example Products | Function in the Workflow |
|---|---|---|
| RNA Isolation Kits | PureLink RNA Mini Kit, MagMAX-96 Total RNA Isolation Kit, mirVana miRNA Isolation Kit, RNAqueous-Micro Kit [29] | Purify high-quality RNA from various sample types (fresh/frozen cells, tissue, FFPE), with options for total RNA or specific RNA-size populations. |
| RNA Quantification Assays | Qubit RNA Assay, Quant-iT RNA Assay [29] | Provide specific, sensitive quantification of RNA concentration with minimal interference from common contaminants, superior to UV absorbance. |
| Reverse Transcription Kits | SuperScript IV VILO Master Mix [29] | Generate high-quality first-strand cDNA with reduced amplification bias, available in convenient single-tube formats. |
| qPCR Assays & Plates | TaqMan Gene Expression Assays, TaqMan Array Plates (96- or 384-well), TaqMan OpenArray Plates [9] | Enable highly specific and reproducible quantification of targeted gene expression, available in various throughput formats to suit experimental scale. |
| RNA-seq Library Prep Kits | TruSeq stranded mRNA kit (Illumina), SureSelect XTHS2 RNA kit (Agilent) [31] | Prepare sequencing libraries from RNA, often involving mRNA enrichment, fragmentation, adapter ligation, and index addition. |
| Reference Gene Selection Software | GSV (Gene Selector for Validation) Software [8] | Identifies the most stable and highly expressed reference genes from RNA-seq data (TPM values) for optimal normalization in downstream qPCR validation. |
The question is not whether to use qPCR or RNA-seq, but how to best use them together. The integrated workflow—employing qPCR upstream for quality control of cDNA and downstream for validation of critical findings on independent samples—creates a powerful, self-reinforcing cycle of discovery and verification. This approach maximizes data integrity, increases confidence in results for manuscript publication, and provides a cost-effective strategy for robust transcriptomic analysis in research and drug development. By understanding the distinct strengths and optimal applications of each technology, scientists can design more reliable and impactful gene expression studies.
Within the context of gene expression analysis, the choice between RNA sequencing (RNA-seq) and quantitative PCR (qPCR) is not necessarily an either/or decision. While qPCR remains the gold standard for targeted gene expression quantification due to its wide dynamic range, low quantification limits, and cost-effectiveness for analyzing a limited number of genes, RNA-seq provides an unbiased, genome-scale view of the transcriptome [9] [32]. A critical component of harnessing the power of RNA-seq is the construction of a robust bioinformatics pipeline, the design of which directly impacts the accuracy, reproducibility, and biological validity of the results [33] [34]. This guide objectively compares the performance of common tools and methods for alignment, quantification, and normalization, providing supporting experimental data to inform researchers and drug development professionals designing pipelines for differential expression research.
RNA-seq Workflow and Key Decision Points
A typical RNA-seq data analysis begins with raw sequencing reads and proceeds through a series of preprocessing steps before biological interpretation can occur. The key stages and the choices made at each point significantly influence the downstream results [33].
Key stages and tool choices in the RNA-seq preprocessing pipeline [33].
Experimental Insights from Multi-Center Benchmarking
Large-scale benchmarking studies provide critical empirical data on how pipeline choices affect real-world outcomes. A landmark study involving 45 laboratories, which generated over 120 billion reads from 1080 libraries, systematically evaluated 26 experimental processes and 140 bioinformatics pipelines [34]. This study revealed that each bioinformatics step, as well as experimental factors like mRNA enrichment and library strandedness, are primary sources of variation in gene expression measurements. Notably, inter-laboratory variations were significantly greater when detecting subtle differential expression—a common scenario in clinical diagnostics—compared to large expression differences [34].
Alignment Tools: Performance Comparison
The alignment (or mapping) step involves matching sequencing reads to a reference transcriptome or genome to identify their genomic origin. Researchers can choose between traditional alignment-spliced alignment tools and pseudoalignment methods, which estimate transcript abundances without base-by-base alignment [33].
Table 1: Comparison of RNA-seq Alignment and Quantification Tools
| Tool | Type | Key Features | Considerations | Benchmarking Performance |
|---|---|---|---|---|
| STAR [33] | Spliced Aligner | Aligns non-contiguous reads across introns; precise mapping | Higher computational memory requirements | Widely used; performance varies with library preparation [34] |
| HISAT2 [33] | Spliced Aligner | Memory-efficient; fast alignment using global FM index | Common in benchmarks; influenced by experimental protocol [34] | |
| TopHat2 [33] | Spliced Aligner | Early standard for spliced alignment; uses Bowtie2 | Largely superseded by STAR and HISAT2 | |
| Kallisto [33] [31] | Pseudoaligner | Ultra-fast; uses kallisto index for abundance estimation; bootstrapping for uncertainty | Does not produce base-by-base genomic coordinates | Faster, less memory; accurate for expression estimation [33] |
| Salmon [33] | Pseudoaligner | Fast, bias-corrected quantification; models fragment GC-content bias | Does not produce base-by-base genomic coordinates | Faster, less memory; accurate for expression estimation [33] |
Quantification and Normalization Strategies
Following alignment, reads are assigned to genomic features such as genes or transcripts (quantification), and the resulting counts are adjusted to make samples comparable (normalization).
Read Quantification
Quantification tools generate a raw count matrix, where the number of reads mapped to each gene in each sample is summarized. A higher number of reads indicates higher expression of that gene [33]. Tools like featureCounts and HTSeq-count are commonly used for this purpose when starting from aligned BAM files. Alternatively, pseudoaligners like Kallisto and Salmon perform alignment and quantification simultaneously, outputting transcript abundances directly [33] [31].
Normalization Methods
Normalization is a critical statistical adjustment to remove technical biases, such as differences in sequencing depth between samples. Without proper normalization, samples with more total reads would appear to have higher gene expression across the board, obscuring true biological differences [33].
Table 2: Common RNA-Seq Normalization Methods and Their Applications
| Method | Sequencing Depth Correction | Gene Length Correction | Library Composition Correction | Suitable for DE Analysis | Notes |
|---|---|---|---|---|---|
| CPM (Counts per Million) [33] | Yes | No | No | No | Simple scaling by total reads; highly affected by a few highly expressed genes. |
| RPKM/FPKM [33] | Yes | Yes | No | No | Enables within-sample comparison; not for cross-sample comparison due to composition bias. |
| TPM (Transcripts per Million) [33] | Yes | Yes | Partial | No | Scales sample to a constant total (1 million); good for cross-sample comparison. |
| Median-of-Ratios (DESeq2) [33] | Yes | No | Yes | Yes | Robust to composition bias; assumed majority of genes are not differentially expressed. |
| TMM (Trimmed Mean of M-values, edgeR) [33] | Yes | No | Yes | Yes | Robust to composition bias; similar assumptions to median-of-ratios. |
The choice of normalization method is pivotal for the accuracy of differential gene expression (DGE) analysis. Methods like CPM, FPKM, and TPM are not considered suitable for DGE analysis because they do not adequately correct for library composition biases. In contrast, the median-of-ratios method (used by DESeq2) and the TMM method (used by edgeR) are specifically designed for this purpose and incorporate statistical models that account for inter-sample variability [33].
Experimental Protocols for Benchmarking
Large-Scale Multi-Center Benchmarking Design
The design of the Quartet project provides a robust template for evaluating RNA-seq pipelines [34]:
- Reference Materials: Four well-characterized RNA samples from a family quartet (parents and monozygotic twins) with small biological differences, spiked with ERCC RNA controls. MAQC samples with larger biological differences were used in parallel.
- Sample Mixing: Two additional samples (T1, T2) were created by mixing two Quartet samples at defined ratios (3:1 and 1:3), providing a built-in truth for ratio-based expression changes.
- Data Generation and Analysis: 45 independent laboratories processed the same sample panel using their in-house experimental protocols and bioinformatics pipelines (totaling 140 distinct pipelines). This design captured real-world technical variation.
- Performance Metrics: Accuracy was assessed against multiple ground truths, including TaqMan qPCR data (for absolute expression), known spike-in concentrations (ERCC), and expected mixing ratios. Reproducibility was measured across technical replicates and laboratories [34].
Protocol for Cross-Study Predictive Performance
To assess how preprocessing affects a pipeline's generalizability, the following protocol can be implemented [35]:
- Dataset Splitting: A large dataset (e.g., TCGA with ~7,870 samples) is split into training (80%) and internal test (20%) sets.
- Independent Validation: The model is tested on completely independent datasets (e.g., GTEx with ~3,340 samples or combined ICGC/GEO with ~876 samples).
- Preprocessing Variations: Different combinations of normalization (e.g., Quantile Normalization), batch effect correction (e.g., ComBat), and data scaling are applied to the training set.
- Model Training and Evaluation: A classifier (e.g., Support Vector Machine) is trained on the preprocessed data and evaluated on the independent test sets. Performance metrics (e.g., F1-score) reveal which preprocessing steps improve cross-study predictions [35].
The Scientist's Toolkit
Table 3: Essential Reagents and Software for RNA-seq Analysis
| Item | Function | Example Products/Tools |
|---|---|---|
| RNA Isolation Kit | Purifies intact, high-quality total RNA from samples. | AllPrep DNA/RNA Mini Kit (Qiagen), AllPrep DNA/RNA FFPE Kit (Qiagen) [31]. |
| Library Prep Kit | Converts RNA into a sequenceable library; often includes mRNA enrichment and adapter ligation. | TruSeq stranded mRNA kit (Illumina), SureSelect XTHS2 RNA kit (Agilent) [31] [36]. |
| Quality Control Software | Assesses raw read quality, adapter contamination, and other potential issues. | FastQC, multiQC, RSeQC [33] [31]. |
| Alignment Software | Maps sequencing reads to a reference genome/transcriptome. | STAR, HISAT2 [33] [31]. |
| Quantification Software | Counts the number of reads mapped to each gene or transcript. | featureCounts, HTSeq-count, Kallisto, Salmon [33] [31]. |
| Differential Expression Tools | Performs statistical analysis to identify genes expressed at different levels between conditions. | DESeq2, edgeR [33]. |
Designing an RNA-seq pipeline requires careful consideration of the trade-offs associated with each tool and method. Large-scale benchmarking studies reveal that the choices for alignment, quantification, and normalization are not merely technical details but are primary sources of variation that can significantly impact results, especially when seeking to identify subtle differential expression [34]. While pseudoalignment tools like Kallisto and Salmon offer speed advantages, traditional aligners like STAR provide genomic mapping. For normalization, methods embedded in dedicated DGE tools like DESeq2 and edgeR are specifically designed for robust cross-sample comparison. The optimal pipeline is often dictated by the specific biological question, the required precision, and the available computational resources. Furthermore, in the broader context of RNA-seq and qPCR, the two methods are frequently complementary; qPCR serves as a valuable independent technique for validating key findings from large-scale RNA-seq screens [9].
Selecting Optimal Reference Genes for qPCR Using RNA-seq Data
Quantitative real-time PCR (qPCR) remains the gold standard technique for validating gene expression data obtained from high-throughput RNA sequencing (RNA-seq) due to its superior sensitivity, specificity, and reproducibility [37]. However, the accuracy of qPCR data critically depends on proper normalization to account for technical variations introduced during RNA isolation, reverse transcription, and amplification [37]. Inadequate normalization can lead to misinterpretation of biological results, with false positives or negatives potentially exceeding 20-fold in extreme cases [37].
The traditional approach utilizes reference genes (RGs)—preferably multiple—that are stably expressed across all experimental conditions [38]. The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines strongly recommend validating reference genes for each specific experimental system [38] [39]. With the exponential growth of publicly available RNA-seq datasets, researchers now have an unprecedented opportunity to mine these resources for identifying optimal reference genes in silico before laboratory validation [40]. This guide comprehensively compares methods for selecting reference genes using RNA-seq data, providing researchers with actionable protocols and performance evaluations.
Methodological Framework: From RNA-seq Data to Validated Reference Genes
RNA-seq Mining for Stable Gene Identification
The foundational approach involves computational mining of RNA-seq datasets to identify genes with stable expression across diverse conditions relevant to your experimental design. This method leverages large-scale transcriptomic data to pre-screen potential reference genes before laboratory validation.
Experimental Protocol:
- Dataset Curation: Compile RNA-seq data encompassing the biological conditions of interest (e.g., tissues, treatments, developmental stages). The tomato study utilized TomExpress database containing 394 biological conditions [40].
- Expression Quantification: Extract normalized expression values (e.g., TPM, FPKM) for all genes across all samples.
- Stability Calculation: Calculate expression stability statistics for each gene, typically focusing on variance or coefficient of variation across samples.
- Candidate Selection: Identify genes with lowest variation, ensuring they have appropriate expression levels (Cq values between 20-30 for optimal qPCR quantification) [40].
Table 1: Performance Comparison of Traditional Housekeeping Genes Versus RNA-seq Identified Genes
| Gene Category | Example Genes | Mean Cq Range | Stability (LVS)* | Tissue Variability |
|---|---|---|---|---|
| Classical HKGs | GAPDH, ACTB, TUB | 18-25 | Variable (0.1-0.9) | High across tissues |
| RNA-seq Identified | IMP-b, RPS18, STAU1 | 20-26 | Consistently high (>0.8) | Low across tissues |
| Low Variance Genes | Ta2776, Ref2, Ta3006 | 22-28 | Highest (>0.95) | Minimal variability |
LVS: Low Variance Score, where 1 represents the most stable gene among those with similar expression levels [40].
The Gene Combination Method
A more sophisticated approach identifies optimal combinations of genes whose expressions balance each other across experimental conditions, even when individual genes show some variability [40]. This method recognizes that a combination of non-stable genes may provide more robust normalization than single stable genes.
Experimental Protocol:
- Target Gene Mean Calculation: Determine the mean expression level of your target gene from RNA-seq data.
- Candidate Pool Selection: Extract the top 500 genes with mean expressions similar to or greater than your target gene [40].
- Combination Optimization: Systematically test all possible geometric and arithmetic combinations of k genes (typically k=2-4) to identify sets where expressions counterbalance.
- Stability Assessment: Select the optimal combination based on two criteria: geometric mean ≥ target gene mean expression, and lowest variance among arithmetic means [40].
Alternative Normalization Strategies
While reference genes remain the most common approach, alternative normalization methods can be preferable in specific scenarios. The global mean (GM) method calculates the average expression of all reliably detected genes in a sample and uses this value for normalization [41]. Algorithm-based approaches like NORMA-Gene use least squares regression to calculate a normalization factor that minimizes technical variation across samples [38].
Table 2: Comparison of Normalization Methods for qPCR Data
| Normalization Method | Minimum Genes Required | Best Application Context | Performance Metrics |
|---|---|---|---|
| Single Reference Gene | 1 | Preliminary studies with limited targets | Variable; high risk of bias |
| Multiple Reference Genes | 2-3 | Standard gene expression studies | CV: 0.275-0.356 [41] |
| Global Mean (GM) | 55+ | High-throughput qPCR (≥55 genes) | Lowest mean CV across tissues [41] |
| NORMA-Gene | 5+ | Studies with no optimal reference genes | Better variance reduction than RGs [38] |
Comparative Performance Analysis
Direct Method Comparison in Experimental Systems
Recent studies have directly compared the performance of different normalization strategies. In canine gastrointestinal tissues, the global mean method outperformed reference gene-based approaches when profiling 81 genes, showing the lowest coefficient of variation across tissues and pathological conditions [41]. In sheep liver studies, NORMA-Gene provided more reliable normalization than traditional reference genes, particularly for oxidative stress genes like GPX3, where interpretation of treatment effects differed significantly between methods [38].
The gene combination method demonstrated particular superiority in tomato studies, where combinations of non-stable genes identified through RNA-seq mining outperformed both classical housekeeping genes and single low-variance genes [40]. This approach recognizes that co-regulated genes with counterbalancing expression patterns can provide more robust normalization than individual stably expressed genes.
Impact on Biological Interpretation
The choice of normalization strategy directly impacts biological conclusions. In wheat studies, normalization using appropriate reference genes (Ref2 and Ta3006) revealed significant differences between absolute and normalized expression values for the TaIPT5 gene across most tissues, while results for TaIPT1 remained consistent regardless of normalization method [42]. This highlights how gene-specific characteristics influence normalization sensitivity.
Diagram 1: RNA-seq to qPCR Reference Gene Pipeline
Implementation Protocols
Laboratory Validation Workflow
After in silico identification of candidate reference genes through RNA-seq mining, rigorous laboratory validation is essential.
Experimental Protocol:
- Primer Design: Design primers with melting temperatures of 57-60°C, GC content of 50-70%, and product sizes of 70-200 base pairs, ensuring they span exon-exon junctions [38].
- Specificity Verification: Confirm primer specificity through sequencing of PCR products and melting curve analysis with single peaks [38] [42].
- Efficiency Calculation: Perform serial dilutions to determine PCR efficiency (ideally 90-110%) using the formula E = 10^(-1/slope) - 1 [41].
- Stability Validation: Analyze candidate gene expression across all experimental conditions using multiple algorithms (geNorm, NormFinder, BestKeeper) [43] [42].
- Comprehensive Ranking: Utilize RefFinder or BruteAggreg to integrate results from all algorithms for final ranking [43] [38].
Troubleshooting Common Issues
Common challenges in reference gene selection include unexpected variability of classical housekeeping genes, insufficient expression stability across conditions, and co-regulation of candidate genes. Ribosomal protein genes frequently show high stability but should not be used exclusively due to potential co-regulation [41]. When traditional reference genes prove unstable, consider algorithm-based approaches like NORMA-Gene or the global mean method, particularly when profiling large gene sets [38] [41].
Diagram 2: Normalization Method Decision Tree
Table 3: Key Research Reagent Solutions for Reference Gene Studies
| Reagent/Resource | Function | Specification Guidelines |
|---|---|---|
| RNA Stabilization | Preserves RNA integrity during storage | RNAlater for tissue preservation [41] |
| RNA Extraction Kits | Isolate high-quality total RNA | RNeasy kits; check RIN/RQI values [38] [37] |
| Reverse Transcriptase | cDNA synthesis from RNA templates | Multiscribe Reverse Transcriptase with random hexamers [38] [44] |
| qPCR Master Mix | Amplification with fluorescence detection | SYBR Green I or TaqMan probes [37] [44] |
| Stability Analysis Software | Reference gene validation | GeNorm, NormFinder, BestKeeper, RefFinder [43] [38] |
| RNA-seq Databases | In silico reference gene mining | TomExpress (plants), GEO (general) [40] |
The integration of RNA-seq data with qPCR experimental design represents a paradigm shift in reference gene selection. Rather than relying on presumed housekeeping genes, researchers can now make evidence-based decisions using comprehensive transcriptomic datasets. The gene combination method emerging from tomato studies demonstrates that optimal normalization may involve multiple genes whose expressions balance each other, rather than individually stable genes [40].
For researchers designing qPCR validation studies, the following evidence-based recommendations are proposed:
- Prioritize RNA-seq Mining: Whenever possible, leverage existing RNA-seq data from your experimental system to pre-screen candidate reference genes in silico [40].
- Implement Combinatorial Approaches: Explore combinations of 2-4 genes identified through RNA-seq analysis, as these often outperform single reference genes [40].
- Validate Extensively: Laboratory validation across all experimental conditions remains essential, using multiple algorithms for comprehensive stability assessment [43] [42].
- Consider Alternatives: For large gene sets (>55 genes), the global mean method provides excellent normalization without requiring reference gene validation [41].
This comparative analysis demonstrates that RNA-seq informed reference gene selection significantly enhances the accuracy and reliability of qPCR data normalization, ultimately strengthening gene expression studies in both basic research and drug development applications.
In the field of gene expression analysis, a fundamental challenge persists: how to reliably interpret and validate differences in gene expression across measurement platforms. The convergence of high-throughput RNA sequencing (RNA-seq) and highly specific quantitative PCR (qPCR) technologies has created a critical need for standardized approaches to assess expression concordance. For researchers in drug development and biomedical research, the accurate identification of differentially expressed genes (DEGs) has direct implications for understanding disease mechanisms, identifying therapeutic targets, and developing biomarkers. This guide examines the core principles of expression concordance assessment, focusing on the complementary roles of correlation coefficients and fold-change measurements in validating transcriptomic data across platforms.
The relationship between RNA-seq and qPCR is inherently synergistic rather than competitive. While RNA-seq provides an unbiased, genome-wide discovery platform, qPCR offers a targeted, highly sensitive validation approach [43] [45]. This complementary relationship necessitates robust methods for cross-platform validation, where correlation statistics quantify the strength of agreement between measurements, and fold-change values assess the magnitude and biological significance of expression differences [46]. Understanding how to interpret these metrics in tandem is essential for establishing confidence in expression findings and ensuring research reproducibility.
Foundational Concepts: Correlation and Fold-Change
Correlation Analysis in Expression Studies
Correlation coefficients, particularly Pearson's r and Spearman's ρ, serve as primary metrics for assessing the technical agreement between RNA-seq and qPCR platforms. These statistics measure how consistently both platforms rank gene expression levels across samples, with values closer to 1.0 indicating perfect agreement. Pearson correlation assesses linear relationships, while Spearman correlation captures monotonic relationships, making the latter more robust to outliers and non-linear amplification effects common in qPCR [47]. High correlation values (typically r > 0.85-0.90) provide confidence that expression patterns detected by RNA-seq reflect true biological signals rather than technical artifacts.
Correlation analysis primarily validates the directional consistency of expression measurements but does not directly address the accuracy of fold-change magnitude. This limitation necessitates complementary analysis using fold-change metrics, particularly for genes with large expression differences where accurate quantification is critical for biological interpretation. The strength of correlation can be influenced by multiple factors including expression level (highly expressed genes typically show better correlation), gene length, and the dynamic range of detection for each platform [47] [17].
Fold-Change as a Biological Significance Metric
Fold-change represents the magnitude of expression difference between conditions and serves as a primary metric for assessing biological significance. In contrast to correlation, fold-change quantification focuses specifically on the effect size that drives biological interpretation. The log2 fold-change (LFC) transformation is standard practice, as it produces symmetric values (e.g., LFC of 1 = 2-fold upregulation, LFC of -1 = 2-fold downregulation) and improves statistical properties for downstream analysis [46].
A critical challenge in fold-change interpretation stems from the systematic differences between platforms. RNA-seq fold-change estimates can be influenced by normalization methods, sequencing depth, and data transformation approaches [46] [48], while qPCR fold-change calculations depend heavily on proper reference gene selection and amplification efficiency corrections [43] [40]. These methodological differences can lead to discrepancies in absolute fold-change magnitude even when directional consistency remains high. Establishing pre-defined thresholds for biological significance (commonly LFC > 1 or 2) helps standardize interpretation across platforms and experimental designs [46].
Quantitative Comparison of RNA-seq and qPCR Platforms
Table 1: Technical Comparison of RNA-seq and qPCR Platforms for Gene Expression Analysis
| Feature | RNA-seq | qPCR |
|---|---|---|
| Throughput | Genome-wide, discovery-oriented [49] | Targeted, validation-focused [45] |
| Dynamic Range | >5 orders of magnitude [47] | 6-8 orders of magnitude [45] |
| Sensitivity | Detects low-abundance transcripts [46] | High sensitivity for rare transcripts [45] |
| Fold-Change Accuracy | Varies by method; DESeq2 recommended for n≥6 [48] | Highly accurate with proper normalization [43] |
| Sample Throughput | High (multiple samples simultaneously) | Medium to high (plate-based) |
| Cost per Sample | Higher | Lower |
| Technical Variability | Moderate; improved with larger sample sizes [49] [48] | Low with technical replicates |
Table 2: Performance of RNA-seq Differential Expression Methods Based on Simulation Studies
| Method | Recommended Sample Size | FDR Control | Power | Stability | Best For |
|---|---|---|---|---|---|
| EBSeq | n = 3 per group [48] | Good | Good | Good | Very small sample sizes |
| DESeq2 | n ≥ 6 per group [48] | Good | Good | Good | Standard experiments |
| edgeR | n ≥ 6 per group [48] | Moderate | Good | Moderate | Standard experiments |
| limma | n ≥ 6 per group [48] | Good | Moderate | Good | Log-normal distributed data |
| SAMSeq | n ≥ 6 per group [49] | Good | Good | Good | Non-parametric analysis |
Experimental Protocols for Cross-Platform Validation
RNA-seq Experimental Workflow
A standardized RNA-seq protocol begins with RNA extraction using high-quality kits that maintain RNA integrity (RIN > 8.0). Library preparation typically employs stranded protocols to maintain transcript orientation information, with sequencing depth recommendations of 20-40 million reads per sample for standard differential expression studies [47]. The computational workflow involves multiple critical steps: quality control (FastQC), adapter trimming (Trimmomatic, Cutadapt), alignment (STAR, HISAT2), quantification (featureCounts, HTSeq), and normalization (TMM, RLE) [47].
For differential expression analysis, method selection should be guided by sample size and data characteristics. Based on comprehensive evaluations, DESeq2 is recommended for studies with at least 6 replicates per group, while EBSeq shows advantages for very small sample sizes (n = 3) [48]. The voom transformation in combination with limma provides robust performance when applying linear models to RNA-seq data [49] [48]. Normalization is critical, with TMM (trimmed mean of M-values) and RLE (relative log expression) methods demonstrating superior performance compared to simple library size normalization [49].
Figure 1: RNA-seq Experimental Workflow: From sample preparation to differential expression analysis.
qPCR Validation Protocol
The qPCR validation workflow begins with careful experimental design, including selection of appropriate reference genes. The MIQE guidelines recommend using at least two validated reference genes for normalization [40] [45]. Reference gene stability should be assessed using multiple algorithms (geNorm, NormFinder, BestKeeper) integrated through RefFinder [43] [45]. RNA extraction and cDNA synthesis protocols must be optimized to minimize degradation and ensure efficient reverse transcription.
For assay design, primers should demonstrate 90-110% amplification efficiency with R² > 0.98 in standard curves [45]. Each reaction should include technical triplicates and no-template controls. Data analysis typically uses the ΔΔCt method for relative quantification, with efficiency correction when necessary [45]. The selection of target genes for validation should include both strongly differentially expressed genes and those with moderate fold-changes to properly assess the dynamic range of concordance.
Figure 2: qPCR Validation Workflow: From experimental design to concordance assessment.
Analytical Framework for Concordance Assessment
Statistical Assessment of Platform Agreement
A robust concordance assessment integrates both correlation and fold-change metrics. The analysis should begin with scatter plots of expression values (log-transformed) from both platforms, with calculation of Pearson and Spearman correlation coefficients. However, correlation alone is insufficient, as it can be inflated by genes with extreme expression values. Bland-Altman analysis (plotting the difference between measurements against their mean) provides additional insight into systematic biases between platforms [47].
For fold-change comparison, scatter plots of LFC values should demonstrate clustering around the y=x line. The concordance correlation coefficient (CCC) integrates both precision (correlation) and accuracy (deviation from y=x) in a single metric. Pre-defined acceptance criteria should be established, such as >85% of validated genes showing directional consistency and >80% showing LFC ratios (RNA-seq/qPCR) between 0.5-2.0 [46] [47]. Statistical significance should be considered alongside effect size, as small expression changes with low p-values may lack biological relevance.
Interpretation Guidelines for Discrepant Results
When discordant results occur between platforms, systematic troubleshooting should investigate both technical and biological factors. Technical factors include RNA quality differences, reference gene instability in qPCR, normalization methods in RNA-seq, and platform-specific biases (e.g., GC content effects) [43] [47]. Biological factors include alternative transcript detection (RNA-seq may detect isoforms not amplified by qPCR primers) and time-dependent expression changes when analyses are conducted separately [17].
Particular attention should be paid to genes with low expression levels, as these demonstrate poorer concordance between platforms. RNA-seq may struggle with accurate quantification of low-count genes, while qPCR may encounter issues with detection limits and higher variability near the Ct threshold [47] [45]. Establishing expression level thresholds (e.g., FPKM > 1 or Ct < 30) for concordance assessment can improve overall agreement metrics by excluding problematic low-abundance transcripts.
Research Reagent Solutions
Table 3: Essential Research Reagents and Tools for Expression Concordance Studies
| Category | Specific Products/Tools | Function | Considerations |
|---|---|---|---|
| RNA Isolation | TRIzol, RNeasy Plus Mini kit [47] [45] | RNA extraction and purification | Maintain RNA integrity (RIN > 8.0) |
| RNA Quality Assessment | Agilent 2100 Bioanalyzer [47] | RNA integrity number (RIN) calculation | Essential for both platforms |
| cDNA Synthesis | SuperScript First-Strand Synthesis System, PrimeScript RT reagent Kit [47] [45] | Reverse transcription of RNA to cDNA | Use random hexamers and oligo-dT |
| qPCR Master Mix | TB Green Premix Ex Taq II [45] | Fluorescence-based detection | SYBR Green or probe-based |
| Reference Genes | arf1, rpL32 (honeybee) [45]; IbACT, IbARF (sweet potato) [43] | Normalization of qPCR data | Must be validated for each experiment |
| RNA-seq Library Prep | TruSeq Stranded Total RNA Kit [47] | Library preparation for sequencing | Maintain strand specificity |
| Alignment Tools | STAR, HISAT2 [47] | Read alignment to reference genome | Spliced alignment required |
| Quantification Tools | featureCounts, HTSeq [47] | Gene-level read counting | Assignment of multimapping reads |
| Differential Expression | DESeq2, edgeR, limma [48] | Statistical analysis of DEGs | Choose based on sample size |
The assessment of expression concordance between RNA-seq and qPCR requires integrated interpretation of both correlation and fold-change metrics. Through systematic analysis of comparative studies, several best practices emerge. First, researchers should select RNA-seq analysis methods appropriate for their sample size, with DESeq2 recommended for studies with at least 6 replicates per group [48]. Second, qPCR validation must include properly validated reference genes using stability measures across the specific experimental conditions [43] [45]. Third, concordance thresholds should be established a priori, with expectations for correlation strength and fold-change agreement tailored to expression level ranges.
The evolving landscape of transcriptomics continues to introduce new technologies and analysis methods. Long-read sequencing approaches are improving transcript isoform resolution [17], while more sophisticated normalization strategies are enhancing cross-platform comparability. Nevertheless, the fundamental framework of assessing both correlation (direction consistency) and fold-change (magnitude agreement) remains essential for rigorous validation of gene expression findings. By implementing the standardized protocols and analytical approaches outlined in this guide, researchers can enhance the reliability and reproducibility of their expression studies, ultimately strengthening the translational potential of their discoveries in drug development and clinical applications.
Optimizing Accuracy and Overcoming Technical Pitfalls in Both Methods
Quantitative PCR (qPCR) remains a cornerstone technique for gene expression analysis, despite the rising adoption of RNA sequencing (RNA-seq) [50] [51]. Its role in validating differential expression findings from RNA-seq is particularly critical, as it provides independent, highly precise quantification of specific transcripts [3] [52]. However, researchers frequently encounter technical challenges that can compromise data quality and reproducibility. Issues such as low yield, non-specific amplification, and variation in quantification cycle (Cq) values can obscure true biological signals and lead to erroneous conclusions in both basic research and drug development [50] [53]. This guide objectively examines these common qPCR pitfalls, provides troubleshooting strategies grounded in experimental data, and contrasts qPCR's targeted approach with the broader discovery potential of RNA-seq. By understanding these technical challenges and their solutions, researchers can ensure their gene expression data, whether generated for initial discovery or final validation, is robust and reliable.
Understanding and Troubleshooting Common qPCR Challenges
Low Yield
Low yield in qPCR refers to suboptimal reaction efficiency, resulting in less quantitative data and reduced sensitivity [50]. This issue often manifests as unexpectedly high Cq values (indicating low initial template concentration) or a low fluorescence plateau.
- Primary Causes and Solutions:
- RNA Quality: Degraded RNA or the presence of inhibitors can severely impact yield [50]. Ensure high-integrity RNA by using optimized purification steps and appropriate clean-up procedures [50].
- cDNA Synthesis: Inefficient reverse transcription is a common bottleneck. Adjust cDNA synthesis conditions and ensure consistent reagent volumes to improve efficiency [50].
- Primer Design: Suboptimal primers are a frequent culprit. Use specialized design software to create primers with appropriate length, GC content (typically 30-50%), and melting temperature (Tm), while checking for potential secondary structures [50] [54].
Non-Specific Amplification
Non-specific amplification occurs when primers anneal to non-target sequences, leading to the amplification of unintended products or primer-dimers. This can result in overestimated template concentration and inaccurate Cq values [50] [54].
- Primary Causes and Solutions:
- Primer Design: Redesign primers using specialized software to avoid primer-dimers and ensure target specificity [50]. Aim for primer melting temperatures within 2-5°C of each other [54].
- Annealing Temperature: An annealing temperature that is too low is a common cause. Optimize the annealing temperature by running a gradient PCR to determine the temperature that maximizes specific product yield while minimizing non-specific products [50] [54].
- Reaction Components: Optimize primer and Mg²⁺ concentrations, as these can influence primer specificity and amplification fidelity [55].
Cq (Ct) Value Variations
The quantification cycle (Cq), also known as Ct, is the cycle number at which the fluorescence signal crosses a defined threshold [53] [56]. Unexplained variations in Cq values between technical replicates or expected samples can compromise data integrity and interpretation.
- Primary Causes and Solutions:
- Pipetting Errors: Inconsistent pipetting is a major source of technical variation. Ensure proper pipetting techniques, calibrate pipettes regularly, and use positive-displacement pipettes with filtered tips for small volumes [50] [54]. Automated liquid handlers can significantly improve reproducibility [50].
- Reaction Efficiency: Differences in PCR efficiency greater than 5% between samples can cause significant Cq variations [53] [55]. Calculate reaction efficiency from standard curves and redesign assays if efficiency is below 88% or differs substantially between samples [54] [55].
- Inhibitors and Contamination: Sample contaminants or genomic DNA contamination (in RT-qPCR) can affect amplification. DNAse-treat RNA samples, dilute samples to reduce inhibitor concentration, and maintain a clean pre-PCR workspace [54].
The workflow below illustrates the logical process for diagnosing and addressing these common qPCR issues.
qPCR vs. RNA-seq: A Data-Driven Comparison for Expression Analysis
When designing validation studies, understanding the complementary strengths and limitations of qPCR and RNA-seq is crucial. The table below summarizes their core characteristics.
Table 1: Comparison of qPCR and RNA-seq for Gene Expression Analysis
| Feature | qPCR | RNA-seq (Transcriptome-Wide) | Targeted RNA-seq |
|---|---|---|---|
| Throughput | Low to medium (1-10s of targets) [52] | High (entire transcriptome) [52] | Medium (predefined gene sets) [52] |
| Dynamic Range | ~7-8 logs [57] | >5 logs [52] | >5 logs [52] |
| Accuracy & Precision | High for known targets [52] | High, but can be influenced by alignment biases [3] | High for selected targets [52] |
| Primary Application | Targeted validation, hypothesis-driven research [52] | Discovery, novel transcript identification [52] | Focused profiling, clinical panels [52] |
| Sample Quality Requirement | High-quality RNA often required | High-quality RNA for full transcriptome; degraded RNA (e.g., FFPE) can be used with specific protocols [52] | More tolerant of degraded RNA [52] |
| Cost per Sample | Low [52] | High [52] | Moderate [52] |
| Time to Result | 1-3 days [52] | Several days to weeks (includes bioinformatics) [52] | Several days [52] |
| Ease of Data Analysis | Relatively simple, requires minimal bioinformatics [52] | Complex, requires significant bioinformatics expertise [3] [52] | Moderate, streamlined bioinformatics [52] |
A direct experimental comparison highlights the challenges in correlating results from these two technologies. A 2023 study comparing HLA class I gene expression measured by both qPCR and RNA-seq in the same samples found only a moderate correlation, with Pearson correlation coefficients (rho) ranging from 0.2 to 0.53 for HLA-A, -B, and -C [3]. This discrepancy underscores the technical and biological factors differentiating these methods, such as:
- Alignment Biases in RNA-seq: The extreme polymorphism of genes like HLA complicates the alignment of short reads to a reference genome, potentially leading to inaccurate quantification [3].
- Normalization Differences: qPCR typically uses a small set of reference genes for normalization, while RNA-seq relies on global normalization methods, which can yield different expression estimates [55].
Therefore, while qPCR is excellent for validating the direction and magnitude of expression changes for a small number of key genes identified in an RNA-seq screen, the absolute values from these different techniques may not be directly interchangeable.
Experimental Protocols for Robust qPCR Assays
Protocol for Primer Validation and Efficiency Calculation
A critical step in any qPCR experiment is validating the performance of the primer sets [57].
- Serial Dilution: Prepare a logarithmic dilution series (e.g., 1:10 dilutions) of a sample containing the target, spanning at least 3 orders of magnitude [55] [57].
- qPCR Run: Amplify each dilution in triplicate, including a no-template control (NTC).
- Standard Curve: Plot the mean Cq value for each dilution against the logarithm of its concentration. Perform linear regression on the data points [57].
- Efficiency Calculation: Calculate the amplification efficiency (E) using the slope of the standard curve: E = 10^(-1/slope) - 1 [55] [57].
- Linearity Assessment: The coefficient of determination (R²) for the standard curve should be ≥ 0.98, indicating a strong linear relationship [57].
Protocol for Distinguishing Specific from Non-Specific Amplification
Using intercalating dye chemistry (e.g., SYBR Green I) requires confirming amplification specificity.
- Melt Curve Analysis: After the final PCR cycle, slowly heat the amplicons from around 60°C to 95°C while continuously monitoring fluorescence. A sharp, single peak indicates a single, specific PCR product. Multiple peaks or a broad peak suggest primer-dimers or non-specific products [57].
- Gel Electrophoresis: Run the qPCR products on an agarose gel. A single band of the expected size confirms specific amplification, while multiple bands or a smear indicate non-specificity [57].
The Scientist's Toolkit: Essential Reagents and Materials
Successful qPCR relies on high-quality reagents and conscientious techniques. The following table lists key solutions and their critical functions in the workflow.
Table 2: Essential Reagents and Materials for a Robust qPCR Workflow
| Item | Function | Key Considerations |
|---|---|---|
| High-Quality RNA Isolation Kit | To obtain pure, intact RNA free of genomic DNA and inhibitors. | Include a DNase I digestion step; assess RNA Integrity Number (RIN) or via gel electrophoresis [3] [58]. |
| Reverse Transcriptase | Synthesizes cDNA from RNA templates. | Use a robust enzyme; avoid multiple freeze-thaw cycles to prevent degradation [54]. |
| qPCR Master Mix | Provides polymerase, dNTPs, buffer, and fluorescence dye/probe. | Use high-quality, consistent mixes; check for desired sensitivity and compatible chemistry (SYBR Green vs. Probe) [54] [57]. |
| Sequence-Specific Primers | Amplify the target of interest. | Designed for specificity, optimal Tm (~60°C), and lack of secondary structures; validate efficiency [50] [57]. |
| Passive Reference Dye (e.g., ROX) | Normalizes for non-PCR-related fluorescence fluctuations between wells. | Ensures consistent dye concentration across reactions for accurate fluorescence readings [54]. |
| Nuclease-Free Water | Solvent for dilutions and master mix preparation. | Guaranteed free of nucleases and contaminants that could degrade samples or inhibit reactions. |
| Optical Plates & Seals | Vessel for the qPCR reaction. | Must be optically clear for fluorescence detection and provide a tight seal to prevent evaporation. |
Navigating the common challenges of low yield, non-specific amplification, and Cq variation is fundamental to generating reliable qPCR data. As demonstrated, these issues have well-defined causes and solutions, ranging from rigorous primer design and validation to meticulous pipetting and the use of automation [50] [54]. In the context of a research pipeline that may also employ RNA-seq, qPCR's enduring value lies in its precision, sensitivity, and cost-effectiveness for targeted gene validation [52]. By adhering to optimized protocols, understanding the technology's limitations, and following established guidelines like MIQE [51] [57], researchers can confidently use qPCR to produce publication-quality results that robustly support their findings in differential expression studies.
Primer Design and Concentration Fine-Tuning for Robust qPCR Efficiency
In the evolving landscape of gene expression analysis, quantitative PCR (qPCR) maintains a critical role as a validation tool for high-throughput technologies like RNA-sequencing (RNA-seq). While RNA-seq has become the gold standard for whole-transcriptome profiling [2], qPCR remains indispensable for confirming differential expression findings with higher sensitivity and precision [3]. The reliability of qPCR data, however, fundamentally depends on robust assay design and optimization, particularly through careful primer design and concentration fine-tuning. Within the context of RNA-seq versus qPCR validation studies, proper primer design becomes not merely a technical prerequisite but a cornerstone for generating comparable, reproducible data across platforms. This guide systematically compares optimization strategies to achieve superior qPCR efficiency, providing researchers with practical methodologies to bridge technological divides in gene expression research.
Foundational Principles of qPCR Primer Design
Effective primer design requires balancing multiple parameters to ensure specific and efficient amplification. The core principles governing this process have been established through extensive empirical research and are implemented in various bioinformatics tools.
Core Design Parameters
Length and Melting Temperature (Tm): Primers should typically be 18–30 nucleotides in length to ensure specificity while maintaining an adequate Tm [59] [60]. The optimal melting temperature for primers is 60–64°C, with both primers in a pair having Tm values within 2°C of each other to synchronize annealing [59]. Tm calculation should account for specific reaction conditions, including cation concentrations, using tools like the IDT OligoAnalyzer or NEB's Tm calculator [59] [60].
GC Content and Sequence Composition: Ideal primer GC content falls between 40–60%, providing stable binding without promoting secondary structures [59] [61]. Sequences should avoid regions of 4 or more consecutive G residues, and a guanine base at the 5' end of probes should be avoided as it can quench fluorophore emission [59] [60].
Specificity and Secondary Structures: Primers must be screened for self-dimers, heterodimers, and hairpin formation. The free energy (ΔG) for any such structures should be weaker (more positive) than -9.0 kcal/mol [59]. Using alignment tools like NCBI BLAST is essential for verifying primer uniqueness to the intended target [59] [62].
Advanced Design Considerations
Amplicon Characteristics: Target amplicon lengths of 70–200 base pairs maximize PCR efficiency [60]. For gene expression studies using cDNA, designs should span an exon-exon junction where possible to prevent amplification of contaminating genomic DNA [59] [62] [60].
Probe Design for Hydrolysis Assays: When using TaqMan probes, the probe Tm should be 5–10°C higher than the primer Tm to ensure the probe binds before the primers [59] [60]. Probes are typically 15–30 nucleotides long, with optimal concentrations between 100–500 nM [60]. Double-quenched probes are recommended over single-quenched probes for lower background and higher signal [59].
Comparative Analysis of Primer Design and Optimization Tools
Researchers have access to numerous bioinformatics tools for primer design, each with distinct strengths and specializations. The table below summarizes key available tools and their primary applications.
Table 1: Comparison of qPCR Primer and Probe Design Tools
| Tool Name | Provider | Key Features | Best Suited For |
|---|---|---|---|
| PrimerQuest | IDT | Customizes ~45 parameters; designs primers, probes for intercalating dyes or hydrolysis assays [63]. | Researchers requiring highly customized assay designs with specific parameters. |
| NCBI Primer-BLAST | NIH/NLM | Integrates primer specificity checking directly against sequence databases; options to span exon junctions [62]. | Ensuring absolute primer specificity and designing gene-specific assays. |
| RealTime qPCR Design Tool | IDT | Designs assays across exon boundaries for non-standard species [59]. | Working with gene targets in species other than human, mouse, and rat. |
| TaqMan Design Tool | GenScript | Automated design of primers and probes for hydrolysis assays; can define exon junctions manually [64]. | Quick design of TaqMan-style assays with minimal user input. |
| OligoAnalyzer Tool | IDT | Analyzes Tm, hairpins, dimers, and off-target binding via BLAST [59]. | Rapid analysis and troubleshooting of pre-designed oligonucleotides. |
Experimental Protocols for Concentration Fine-Tuning
After in silico design, empirical optimization of primer concentrations and cycling conditions is essential for developing a robust qPCR assay. Indications of poor optimization include lack of reproducibility between replicates, inefficient amplification, and insensitive assays [65].
Annealing Temperature Optimization Using a Gradient
Determining the optimal annealing temperature (Ta) is a critical first step in assay optimization, which can be efficiently achieved using a thermocycler with a gradient block function.
Table 2: Reaction Setup for Annealing Temperature Optimization
| Component | Volume per 20 µL Reaction | Final Concentration |
|---|---|---|
| SYBR Green ReadyMix (2X) | 10 µL | 1X |
| Forward Primer (10 µM) | 0.5 µL | 450 nM * |
| Reverse Primer (10 µM) | 0.5 µL | 450 nM * |
| Template (cDNA/gDNA) | Variable | Diluted 1:5-1:10 [65] |
| PCR-grade Water | To 20 µL | - |
*Primer concentration can be used as a starting point for optimization [65].
Protocol:
- Prepare Master Mix: Create a master mix sufficient for all reactions, including no-template controls (NTCs) [65].
- Plate Setup: Aliquot the master mix into a qPCR plate. Using a temperature gradient instrument, set up identical reactions across a temperature range (e.g., 54–70°C) [65].
- Run qPCR Program: Use a standard three-step protocol with 40 cycles. The annealing temperature should be defined as a gradient across the plate [65].
- Analysis: The optimal Ta is the highest temperature that yields the lowest Cq value and highest amplification efficiency, typically no more than 5°C below the Tm of the primers [59].
Primer Concentration Titration
Once the optimal Ta is established, fine-tuning primer concentration can further enhance specificity and signal strength.
- Test a Concentration Series: Prepare reactions with primer concentrations ranging from 100–500 nM for dye-based qPCR and 200–900 nM for probe-based assays [60].
- Evaluate Performance: Compare Cq values, amplification efficiency, and endpoint fluorescence across the concentration series. Higher concentrations may increase spurious amplification, while lower concentrations can reduce signal [60].
- Select Optimal Concentration: Choose the concentration that provides the lowest Cq (indicating high efficiency) with a single, specific peak in the melt curve (for SYBR Green) or minimal background fluorescence.
The following workflow diagram summarizes the key stages of the qPCR optimization process.
qPCR Validation in RNA-seq Studies: A Technical Perspective
The relationship between RNA-seq and qPCR is synergistic, with each technology offering complementary strengths. RNA-seq provides an unbiased, genome-wide view of the transcriptome, while qPCR delivers highly accurate, sensitive quantification of a limited number of targets [2]. This makes qPCR the method of choice for validating RNA-seq results.
Correlation and Discordance Between Platforms
Benchmarking studies reveal a high overall concordance between the two platforms. A comprehensive comparison using whole-transcriptome RT-qPCR data for over 18,000 genes found high expression correlations between RNA-seq and qPCR (e.g., R² = 0.845 for Salmon) and high fold-change correlations for differential expression (R² ≈ 0.93 across various workflows) [2]. However, discordance does occur. Approximately 85–90% of genes show consistent differential expression calls between RNA-seq and qPCR, leaving a 10–15% non-concordant fraction [2]. Genes with inconsistent measurements between technologies tend to be shorter, have fewer exons, and are lower expressed [2]. This highlights that validation is particularly crucial for this specific gene set.
The Critical Importance of qPCR Efficiency in Validation
For qPCR to serve as a reliable validation tool, its assays must be highly efficient. The precision of qPCR efficiency estimation directly impacts the accuracy of gene expression ratios. A novel "Pairwise Efficiency" method for analyzing dilution series data has been shown to nearly double the precision of efficiency determination and provide a 2.3-fold improvement in the precision of gene expression ratio estimation compared to the standard calibration curve method [66]. This enhanced precision is vital when confirming subtle but biologically significant fold-changes identified in RNA-seq experiments.
The Scientist's Toolkit: Essential Reagents for qPCR Optimization
Successful qPCR optimization relies on a suite of carefully selected reagents and tools. The table below details key components for establishing a reliable workflow.
Table 3: Research Reagent Solutions for qPCR Optimization
| Item | Function/Role | Specifications & Notes |
|---|---|---|
| Hot-Start DNA Polymerase | Enzyme for DNA amplification; "Hot-Start" reduces non-specific amplification prior to cycling. | Found in commercial master mixes (e.g., Luna Universal qPCR Mix). Ensures specificity [60]. |
| SYBR Green or Hydrolysis Probes | Detection chemistry for quantifying amplification. SYBR Green binds dsDNA; probes offer higher specificity. | Double-quenched probes are recommended for lower background [59]. |
| dNTPs | Nucleotides (dATP, dCTP, dGTP, dTTP) for DNA strand synthesis. | Provided in optimized concentrations in commercial master mixes. |
| Optimal Primers | Bind specifically to the target sequence to initiate amplification. | Designed per guidelines in Section 2; HPLC-purified for best results. |
| Nuclease-Free Water | Solvent for reactions and dilutions; free of RNases and DNases. | Essential for maintaining reagent stability and preventing sample degradation. |
| No-RT & No-Template Controls (NTC) | Critical controls for contamination. No-RT detects gDNA in cDNA preps; NTC detects reagent contamination. | Must be included in every run to ensure data integrity [60]. |
| UDG Treatment | Enzyme to prevent carry-over contamination by degrading uracil-containing DNA from previous runs. | Use 0.025 units/µl Antarctic Thermolabile UDG for 10 min at room temperature [60]. |
| Passive Reference Dye (e.g., ROX) | Normalizes for non-PCR-related fluorescence fluctuations between wells. | Included in many master mixes for broad instrument compatibility [60]. |
Meticulous primer design and systematic concentration optimization are non-negotiable prerequisites for generating publication-quality qPCR data, especially when used to validate RNA-seq findings. By adhering to the detailed guidelines for primer parameters, employing strategic experimental protocols for temperature and concentration fine-tuning, and understanding the technological context of RNA-seq correlation, researchers can achieve robust qPCR efficiencies of 90–110%. This level of precision ensures that qPCR remains a powerful and reliable tool for confirming differential expression, thereby strengthening the conclusions drawn from high-throughput transcriptomic studies.
A technical guide for researchers navigating the complexities of modern transcriptome analysis.
In the field of gene expression analysis, RNA sequencing (RNA-seq) is often contrasted with quantitative PCR (qPCR) for validation of differential expression. While RNA-seq offers an unparalleled, hypothesis-free view of the transcriptome, its accuracy is challenged by technical artifacts that qPCR, as a targeted method, largely avoids. This guide objectively compares the performance of these technologies, focusing on how polymorphisms and alignment difficulties introduce bias into RNA-seq data and providing actionable experimental protocols to mitigate them.
RNA-seq has become the gold standard for whole-transcriptome gene expression quantification, providing an unbiased platform for discovering novel transcripts, alternative splicing, and genetic variants [2]. However, its workflow is extremely intricate, and biases can be introduced at virtually every stage—from sample preservation through library preparation to bioinformatic analysis [67]. These biases can compromise data integrity, leading to erroneous biological interpretations.
A significant source of bias stems from the extreme polymorphism of key immune genes like the Human Leukocyte Antigen (HLA) class I and II loci. Traditional RNA-seq alignment, which maps reads to a single reference genome, struggles with this diversity. Reads carrying alleles not present in the reference may fail to align or align incorrectly, directly impacting expression estimates for these critical genes [3]. This article will dissect these challenges and provide a structured comparison with qPCR validation data.
Quantitative Comparison: RNA-seq vs. qPCR
To objectively evaluate performance, we summarize key experimental data from benchmarking studies that compare RNA-seq workflows against whole-transcriptome qPCR data.
Table 1: Comparison of RNA-seq and qPCR Methodologies
| Feature | RNA-seq | qPCR |
|---|---|---|
| Throughput & Discovery | Whole transcriptome; discovers novel transcripts & variants [68] | Limited to known, pre-defined targets (typically ≤ 30 genes) [32] |
| Dynamic Range | Broad [68] | Widest dynamic range and lowest quantification limits for a few targets [32] |
| Key Technical Biases | Alignment errors due to polymorphisms; GC content; library amplification; sequence context [3] [67] | Primer efficiency; amplicon secondary structure |
| Cost & Accessibility | Higher cost; requires significant computing power & bioinformatics expertise [32] | Lower cost for few targets; accessible equipment in most labs [32] [68] |
| Ideal Use Case | Exploratory, discovery-driven research with no reference genome limitations [68] | Targeted validation of a small number of genes with known sequences [32] |
Table 2: Performance Metrics of RNA-seq Workflows vs. qPCR Benchmark
| RNA-seq Workflow | Expression Correlation (R² with qPCR) | Fold-Change Correlation (R² with qPCR) | Fraction of Non-Concordant DE Genes* |
|---|---|---|---|
| Salmon | 0.845 | 0.929 | 19.4% |
| Kallisto | 0.839 | 0.930 | 17.8% |
| Tophat-HTSeq | 0.827 | 0.934 | 15.1% |
| STAR-HTSeq | 0.821 | 0.933 | 15.3% |
| Tophat-Cufflinks | 0.798 | 0.927 | 18.2% |
Non-concordant genes are those for which RNA-seq and qPCR disagree on differential expression status (log fold change > 1). Data adapted from benchmarking against MAQCA/MAQCB samples with 18,080 qPCR assays [2].
Experimental Protocols for Bias Assessment
To ensure the reliability of your RNA-seq data, incorporating the following quality control protocols is essential.
Protocol 1: Assessing Alignment Quality and Genomic Origin
After aligning reads with a splice-aware aligner like STAR, use the aligner’s output log to evaluate basic mapping statistics [69].
- Procedure: Examine the
Log.final.outfile. A uniquely mapping rate of at least 75% is generally good for human/mouse data, while rates below 60% warrant troubleshooting. - Key Metrics: Note the percentages of uniquely mapped reads, reads mapped to multiple loci, and unmapped reads. An unusually high number of multi-mapping reads can indicate issues with repetitive regions or a poorly assembled genome [69].
Following alignment, use tools like RSeQC or Picard's CollectRnaSeqMetrics to determine the genomic distribution of mapped reads [69] [70].
- Procedure: Provide your sorted BAM file and a reference annotation file (in
refFlatformat) to the tool. - Key Metrics:
- Transcript Coverage and 5'-3' Bias: This bias results from errors introduced by reverse transcriptase enzymes during library construction. The tool calculates the ratio of coverage at the 5' end of transcripts to the 3' end [71].
- Reads Genomic Origin: Examine the fraction of reads mapping to exonic, intronic, and intergenic regions. A high intronic mapping rate (>30%) suggests potential genomic DNA contamination or an abundance of pre-mRNA [69].
- Ribosomal RNA (rRNA) Content: Even with depletion, some rRNA remains. Excess ribosomal content (>2%) can be flagged for filtering [69].
Protocol 2: Validating Expression with qPCR
Given the biases revealed in Table 2, validating key findings with qPCR is a critical step.
- Target Selection: Select genes central to your research hypothesis. Prioritize genes that fall into the "non-concordant" category from RNA-seq analysis, as these are most prone to technical discrepancies [2].
- Experimental Design: Follow the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines to ensure reliability and repeatability. This includes designing efficient primers, including appropriate controls, and checking PCR efficiency [32].
- Data Correlation: Compare the log fold changes of your selected genes between the RNA-seq data and the qPCR results. High fold-change correlation (R² > 0.93, as seen in benchmarks) strengthens the credibility of your differential expression findings [2].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Software for Robust RNA-seq Analysis
| Item | Function | Example Products/Tools |
|---|---|---|
| rRNA Depletion Kits | Removes abundant ribosomal RNA, enriching for mRNA and non-coding RNAs. | Illumina Ribo-Zero Plus, QIAseq FastSelect |
| Stranded mRNA Prep Kits | Preserves strand orientation during library construction, crucial for accurate transcript assignment. | Illumina Stranded mRNA Prep [68] |
| Splice-Aware Aligner | Aligns RNA-seq reads across exon-exon junctions to a reference genome. | STAR [69] [70], HISAT2 [70], TopHat2 [70] |
| Pseudoaligner | Provides ultra-fast transcript-level quantification by mapping reads to a transcriptome (not genome). | Salmon [2] [70], Kallisto [2] [70] |
| Variant Caller (RNA-seq Optimized) | Identifies single nucleotide variants (SNVs) and indels from RNA-seq data, accounting for splicing and other artifacts. | VarRNA [72], GATK HaplotypeCaller (with RNA-seq best practices) [72] |
| Alignment QC Tools | Generates comprehensive reports on alignment quality, including genomic origin and coverage uniformity. | RSeQC [70], Qualimap [69], Picard CollectRnaSeqMetrics [71] [69] |
Visualization of RNA-seq Alignment Challenges and Solutions
The following diagram illustrates the core problem of reference bias in RNA-seq alignment and the principle of personalized alignment solutions that help alleviate it.
The data clearly shows that while RNA-seq and qPCR show strong overall correlation, a significant fraction of genes (15-20%) can show inconsistent results between the technologies [2]. These discrepancies are not random but are often systematic, associated with specific gene characteristics like lower expression levels and smaller transcript size [2]. Furthermore, genes within highly polymorphic regions, such as the HLA locus, are particularly susceptible to RNA-seq quantification biases due to alignment challenges [3].
The future of mitigating RNA-seq biases lies in the development and adoption of more sophisticated bioinformatic methods. Tools like iMapSplice, which use personalized genomic information to create unbiased alignment indices, show great promise in alleviating reference bias and improving the detection of personal splice junctions [73]. Similarly, machine learning models like VarRNA are enhancing our ability to accurately call and classify variants directly from RNA-seq data, providing deeper insights into allele-specific expression dynamics in diseases like cancer [72]. For any critical differential expression finding, especially for genes prone to these technical artifacts, validation with the highly specific and sensitive qPCR method remains a cornerstone of rigorous transcriptomics research [32].
Automation and Best Practices to Enhance Reproducibility and Throughput
In the field of transcriptomics, quantitative PCR (qPCR) has long been considered the gold standard for validating differentially expressed genes due to its simplicity, accuracy, and low cost [7]. However, its reliability is heavily dependent on the use of appropriately validated reference genes for normalization, as traditional housekeeping genes can exhibit surprising expression variance across different conditions or species [7]. In contrast, RNA sequencing (RNA-seq) provides a powerful, unbiased approach that enables simultaneous transcriptome-wide discovery and quantification without prior knowledge of gene sequences [74]. As research increasingly demands higher throughput and greater reproducibility, automation and standardized best practices have become crucial for maximizing the potential of both technologies, particularly in drug development contexts where reliable identification of gene expression changes can inform therapeutic target discovery.
This guide objectively compares automated and manual approaches to RNA-seq and qPCR validation, providing experimental data and detailed methodologies to help researchers select appropriate strategies for their differential expression studies.
Technology Comparison: RNA-seq Versus qPCR
Performance Metrics and Experimental Data
Table 1: Comparative Performance of RNA-seq and qPCR for Differential Expression Analysis
| Performance Metric | RNA-seq | qPCR |
|---|---|---|
| Throughput | High (entire transcriptome) | Low to medium (dozens to hundreds of genes) |
| Sensitivity | Detects low-expressed genes [75] | Excellent for detecting even very low abundance transcripts |
| Technical Reproducibility | Highly replicable (Spearman correlation = 0.96 across lanes) [75] | High, but dependent on reference gene stability |
| Differential Expression Detection | Identified 30% more differentially expressed genes than microarrays at same FDR [75] | Considered gold standard when properly normalized |
| Multiplexing Capability | inherently multiplexed | Limited without specialized approaches |
| Reference Dependency | Can be reference-free via de novo assembly [74] | Requires pre-identified reference genes |
| Additional Discoveries | Identifies novel transcripts, splice variants, low-expressed genes [75] | Limited to pre-selected targets |
| Automation Potential | High (demonstrated for library prep and analysis) [76] | Medium (well-established for reaction setup) |
Experimental Evidence for RNA-seq Performance
Technical reproducibility of RNA-seq was rigorously assessed in a landmark study comparing liver and kidney RNA samples sequenced across multiple Illumina lanes. The research demonstrated remarkably high replicability with an average Spearman correlation of 0.96 across technical replicates, suggesting that for many applications, sequencing each mRNA sample in a single lane may suffice [75]. The variation across technical replicates was well-captured using a Poisson model, with only approximately 0.5% of genes showing clear deviations from this model [75].
When comparing RNA-seq's ability to identify differentially expressed genes against established array technologies, the sequence data identified 30% more differentially expressed genes than were obtained from a standard analysis of array data at the same false discovery rate [75]. This enhanced detection power, combined with the ability to identify alternative-spliced forms and novel transcripts, positions RNA-seq as a powerful discovery tool that can effectively complement qPCR's validation strengths.
Automated RNA-seq Workflows for Enhanced Reproducibility
Automated High-Throughput RNA-seq Protocol
Recent advances have demonstrated the feasibility of fully automated RNA-seq workflows integrated with liquid handling stations. The following protocol was successfully implemented for transcriptomic profiling of Saccharomyces cerevisiae under different carbon source conditions [76]:
Table 2: Automated RNA-seq Sample Preparation Workflow
| Step | Process | Key Features | Throughput |
|---|---|---|---|
| Cell Disruption | Enzymatic cell lysis performed at-line | Compatible with difficult-to-lyse samples | 24 samples parallel processing |
| RNA Extraction | Total RNA extraction on liquid handling deck | Integration with purification systems | 24 samples in parallel |
| Library Preparation | Nanopore cDNA library prep | Automated normalization and reagent mixing | 24 libraries in 11.5 hours |
| Sequencing | Oxford Nanopore MinION | Compact device suitable for automation | 20.97 million classified reads (Q>9) |
| Data Analysis | Differential expression pipeline | Automated alignment and quantification | 4 biological replicates per condition |
This automated approach identified significant differences in transcriptomic profiles when comparing growth with glucose (exponential growth) to growth with pyruvate (stress conditions), allowing identification of 674 downregulated and 709 upregulated genes [76]. The expected biological differences confirmed the method's reliability while demonstrating the substantial throughput advantages of automation.
Automation of Single Microbe RNA-seq
For specialized applications requiring single-cell resolution, novel automated approaches have emerged. The smRandom-seq assay utilizes a droplet-based high-throughput single-microbe RNA-seq method that incorporates [77]:
- Microfluidic encapsulation of individual microbes with barcoded beads
- Random primers for in situ cDNA generation (addressing the poly(A) tail limitation in bacteria)
- CRISPR-based rRNA depletion for mRNA enrichment This automated platform achieved high species specificity (99%), minimal doublet rates (1.6%), reduced rRNA percentage (32%), and sensitive gene detection (median of ~1000 genes per single E. coli) [77].
Automated Single-Microbe RNA-seq Workflow
Best Practices for Enhanced Reproducibility
Experimental Design Considerations
Proper experimental design forms the foundation for reproducible RNA-seq results. Key considerations include [74]:
- Replication: The number of replicates should reflect both technical variability in RNA-seq procedures and biological variability of the system, with statistical power calculations informing replicate numbers for detecting expression differences.
- Sequencing Depth: While optimal depth depends on experimental aims, 5 million mapped reads may suffice for medium to highly expressed genes, while up to 100 million reads enable precise quantification of low-expression transcripts.
- RNA Extraction Protocol: Choice between poly(A) selection (requires high mRNA integrity) versus ribosomal depletion (for degraded samples or bacterial RNA).
- Strand-Specific Libraries: Utilization of strand-preserving protocols to properly analyze antisense or overlapping transcripts.
Quality Control Checkpoints
Implementing rigorous quality control at multiple stages is essential for reproducible results [78] [74]:
Table 3: RNA-seq Quality Control Checkpoints
| Analysis Stage | QC Metrics | Tools | Acceptance Criteria |
|---|---|---|---|
| Raw Reads | Sequence quality, GC content, adapter contamination, duplicated reads | FastQC, Trimmomatic | Phred quality score >30, homogeneous GC content across samples |
| Read Alignment | Percentage of mapped reads (70-90% for human), coverage uniformity, strand specificity | Picard, RSeQC, Qualimap | Check for 3' bias indicating RNA degradation |
| Quantification | GC content bias, gene length biases, biotype composition | -- | rRNA should be <5% for poly(A) selected libraries |
| Post-Alignment | rRNA contamination, sample outliers, batch effects | MultiQC, Principal Component Analysis | Remove outliers with >30% disagreement in key metrics |
Reference Gene Validation Using RNA-seq Data
For qPCR validation, RNA-seq data can systematically identify optimal reference genes, overcoming limitations of traditional housekeeping genes. A whole-transcriptome approach enables [7]:
- Identification of stably expressed genes across specific experimental conditions
- Comparison of coefficient of variation (CV) and fold-change methods for stability assessment
- Selection of reference genes with minimal expression variance in target conditions This approach is particularly valuable for non-model organisms where traditional reference genes haven't been established, and can leverage publicly available transcriptomes as low-cost resources for developing better reference genes.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Solutions for RNA-seq Workflows
| Reagent/Category | Function/Purpose | Examples/Specifications |
|---|---|---|
| RNA Stabilization | Preserves RNA integrity immediately post-collection | Liquid nitrogen, dry-ice ethanol baths, -80°C freezer, commercial stabilization reagents |
| RNA Extraction Kits | Isolate high-quality RNA suitable for sequencing | Method matched to sample type, target RNA, and downstream applications |
| Library Prep Kits | Prepare RNA samples for sequencing | Illumina TruSeq (standard input), Takara Bio SMART-Seq v4 (ultra low input), SMARTer Stranded Total RNA-Seq Kit (strandedness) |
| rRNA Depletion | Remove abundant ribosomal RNA | QIAseq FastSelect (>95% rRNA removal in 14 minutes), CRISPR-based depletion (reduces rRNA to 32%) |
| Automation Reagents | Enable high-throughput processing on liquid handlers | Enzymatic cell lysis reagents, normalized nucleic acid concentrations, barcoded beads for single-cell applications |
| Quality Assessment | Verify RNA and library quality | NanoDrop (260/280 ratio >1.8), Agilent TapeStation (RIN >7), Qubit fluorometer, Bioanalyzer |
| Strand-Specific Chemistry | Preserve strand orientation information | dUTP second strand marking methods, other strand-preserving protocols |
Integrated Analysis Workflow for Differential Expression
Integrated Differential Expression Analysis Workflow
Automation and standardized best practices significantly enhance both reproducibility and throughput in RNA-seq studies and subsequent qPCR validation. RNA-seq provides a powerful discovery platform that enables transcriptome-wide differential expression analysis while simultaneously identifying appropriate reference genes for qPCR normalization. The integration of automated workflows from sample preparation through data analysis, coupled with rigorous quality control checkpoints, establishes a robust framework for reliable gene expression studies in basic research and drug development contexts.
As sequencing technologies continue to evolve and automation becomes more accessible, the synergy between high-throughput RNA-seq discovery and targeted qPCR validation will continue to strengthen, providing researchers with complementary tools for unraveling transcriptional regulation with increasing precision and efficiency.
Benchmarking Performance and Determining the Need for Validation
How Reliable is RNA-seq? Evidence from Large-Scale Benchmarking Studies
RNA sequencing (RNA-seq) has become the predominant method for whole-transcriptome gene expression analysis, valued for its ability to profile transcriptomes without prior knowledge of gene sequences and its broad dynamic range. However, as researchers, particularly those in drug development, increasingly rely on RNA-seq data for critical decision-making, a pressing question remains: how reliable are these measurements, especially when compared to established technologies like quantitative PCR (qPCR)? This guide objectively examines the performance of RNA-seq through the lens of large-scale benchmarking studies, providing experimental data and methodologies to help professionals make informed choices about transcriptome analysis in validation workflows.
Quantitative Performance: RNA-seq vs. qPCR
Large-scale consortium-led studies have systematically compared RNA-seq performance against qPCR, which is often considered the gold standard for gene expression validation. The following tables summarize key quantitative findings from these benchmarking efforts.
Table 1: Correlation of Gene Expression Measurements Between RNA-seq and qPCR
| RNA-seq Analysis Workflow | Expression Correlation (Pearson R²) | Fold-Change Correlation (Pearson R²) | Study |
|---|---|---|---|
| Salmon | 0.845 | 0.929 | [2] |
| Kallisto | 0.839 | 0.930 | [2] |
| STAR-HTSeq | 0.821 | 0.933 | [2] |
| TopHat-HTSeq | 0.827 | 0.934 | [2] |
| TopHat-Cufflinks | 0.798 | 0.927 | [2] |
Table 2: Concordance in Differential Expression Analysis
| Performance Metric | Findings | Study |
|---|---|---|
| Non-concordant Genes | 15.1% - 19.4% of genes showed inconsistent differential expression status between RNA-seq and qPCR | [2] |
| HLA Gene Correlation | Moderate correlation between qPCR and RNA-seq for HLA-A, -B, and -C (0.2 ≤ rho ≤ 0.53) | [3] |
| Absolute Quantification | RNA-seq and microarrays do not provide accurate absolute measurements; gene-specific biases are observed | [79] |
Experimental Protocols in Benchmarking Studies
The MAQC/SEQC Consortium Benchmarking
The MicroArray Quality Control (MAQC) and Sequencing Quality Control (SEQC) consortia have conducted some of the most comprehensive assessments of transcriptome technologies.
- Reference Samples: The studies used well-characterized RNA reference samples: Universal Human Reference RNA (MAQCA/UHRR) and Human Brain Reference RNA (MAQCB/HBRR) [2] [79].
- Sample Mixing: Samples C and D were created by mixing A and B in defined ratios (3:1 and 1:3, respectively), creating built-in truths for differential expression [79].
- Spike-in Controls: Synthetic RNA from the External RNA Control Consortium (ERCC) was spiked into samples at known concentrations to assess quantification accuracy [79].
- Multi-site Design: The same reference materials were distributed to multiple independent laboratories for sequencing using different platforms (Illumina HiSeq, SOLiD, Roche 454) and analysis workflows [79].
- qPCR Validation: Extensive qPCR data were generated using TaqMan assays (843 genes) and PrimePCR (20,801 reactions) to serve as a benchmark [79].
The Quartet Project Protocol
The more recent Quartet project focused on assessing RNA-seq's ability to detect subtle differential expression, which is particularly relevant for clinical applications.
- Quartet Reference Materials: The study used four reference materials derived from immortalized B-lymphoblastoid cell lines from a Chinese quartet family, which have smaller biological differences than the MAQC samples [34].
- Study Design: The project involved 45 independent laboratories, each using their in-house experimental protocols and analysis pipelines, reflecting real-world conditions [34].
- Mixed Samples: T1 and T2 samples were constructed by mixing two reference materials at defined ratios of 3:1 and 1:3, providing additional built-in truths [34].
- Performance Metrics: A comprehensive assessment framework was used, including signal-to-noise ratio based on PCA, accuracy of absolute and relative expression, and differential expression analysis [34].
HLA-Specific Expression Benchmarking
A specialized benchmark focused on the challenges of quantifying highly polymorphic HLA genes.
- Sample Collection: RNA was extracted from freshly isolated peripheral blood mononuclear cells (PBMCs) from 96 healthy blood donors [3].
- Multi-method Approach: Each sample was analyzed using three methods: RNA-seq, qPCR, and cell surface HLA-C expression measurement [3].
- HLA-Tailored Bioinformatics: RNA-seq quantification used specialized pipelines that account for extreme HLA polymorphism, minimizing alignment bias present in standard workflows [3].
Visualizing Benchmarking Approaches
The following diagram illustrates the core experimental design used in large-scale RNA-seq benchmarking studies, particularly the MAQC/SEQC and Quartet projects.
The Scientist's Toolkit: Key Reagents & Materials
Table 3: Essential Research Reagents for RNA-seq Benchmarking
| Reagent/Material | Function in Benchmarking | Examples & Specifications |
|---|---|---|
| Reference RNA Samples | Provides consistent, well-characterized materials for cross-platform comparison | MAQCA/UHRR (10 cell lines), MAQCB/HBRR (brain tissue), Quartet family cell lines [2] [34] |
| Spike-in RNA Controls | Assess quantification accuracy and detection limits | ERCC synthetic RNAs (92 variants) with known concentrations [79] |
| RNA Extraction Kits | Maintains RNA integrity and purity for sequencing | Qiagen RNeasy kits; RIN >7 recommended [3] [80] |
| Library Preparation Kits | Converts RNA to sequence-ready libraries; major source of variability | Stranded vs. unstranded protocols; rRNA depletion vs. poly-A selection [80] |
| qPCR Assays | Provides orthogonal validation of expression measurements | TaqMan assays, PrimePCR; thousands of genes for comprehensive validation [79] |
Critical Technical Considerations
Experimental Factors Affecting Reliability
Multiple technical factors significantly impact RNA-seq reliability and cross-study comparability:
- RNA Quality: RNA Integrity Number (RIN) >7 is generally recommended, though rRNA depletion methods can work better with degraded samples than poly-A selection [80].
- Library Strandedness: Stranded libraries are preferred for preserving transcript orientation information, particularly important for identifying long non-coding RNAs and accurately quantifying overlapping genes [80].
- rRNA Depletion: Ribosomal RNA depletion increases coverage of non-ribosomal genes but introduces variability and potential off-target effects; requires careful consideration based on research goals [80].
- Sequencing Depth: Detection of low-abundance transcripts requires sufficient sequencing depth; 10 million reads detects approximately 20,000 genes, while 100 million reads detects >30,000 genes [79].
Bioinformatics Influence on Results
The choice of bioinformatics pipelines substantially affects RNA-seq outcomes:
- Gene Annotation: The completeness of gene annotations dramatically affects mappable reads: 85.9% to RefSeq vs. 97.1% to AceView in one study [79].
- Alignment Tools: Different alignment tools show high concordance for gene-level quantification but substantial variation in junction discovery [79].
- Normalization Methods: Appropriate normalization is critical for cross-sample comparisons, with method performance depending on data characteristics and study design [34].
Performance Across Sequencing Platforms
Recent benchmarking has expanded to include long-read sequencing technologies, providing insights into platform-specific strengths.
- Short-read RNA-seq: Provides robust gene-level expression estimates but struggles with transcript-level quantification for complex isoforms [17].
- Nanopore Long-read RNA-seq: More robustly identifies major isoforms and enables detection of RNA modifications, but has higher input requirements and cost considerations [17].
- PacBio IsoSeq: Provides high-quality full-length transcript sequencing but at lower throughput compared to other platforms [17].
Large-scale benchmarking studies demonstrate that RNA-seq is highly reliable for differential expression analysis when properly controlled, with strong correlation to qPCR for relative quantification (typically R² > 0.9 for fold-changes). However, absolute quantification remains challenging, and performance depends significantly on experimental protocols, bioinformatics choices, and study design. For clinical applications and drug development requiring detection of subtle expression differences, the Quartet project reference materials and multi-laboratory validation provide essential quality control. Researchers should select protocols based on their specific goals, prioritize RNA quality, use spike-in controls, and validate key findings with qPCR, particularly for studies where subtle expression changes have important implications.
The transition from quantitative PCR (qPCR) to RNA sequencing (RNA-seq) for gene expression analysis represents a significant technological shift in molecular biology. While qPCR remains the gold standard for targeted gene expression validation due to its simplicity, accuracy, and low cost, RNA-seq provides an unbiased, genome-wide view of the transcriptome [2] [8]. This comparison guide objectively evaluates the performance concordance between these technologies, examining the factors that contribute to divergent results and providing frameworks for robust experimental design. Understanding the technical underpinnings and limitations of each method is crucial for researchers, scientists, and drug development professionals who rely on accurate gene expression quantification for basic research, biomarker discovery, and clinical applications.
The fundamental differences between these technologies necessitate careful methodological considerations. RNA-seq involves complex bioinformatic processing including read alignment, mapping, and normalization, whereas qPCR depends on primer efficiency and reference gene stability [3] [8]. These procedural differences can lead to variations in gene expression measurements, particularly for challenging genomic regions such as highly polymorphic HLA genes or repetitive elements [3] [81]. Through a systematic analysis of comparative studies and experimental data, this guide provides a comprehensive framework for interpreting concordance and discordance between RNA-seq and qPCR platforms.
Quantitative Comparison of Performance Metrics
Multiple independent studies have systematically compared RNA-seq and qPCR performance using standardized reference samples. The overall correlation between platforms is generally high, though specific performance varies by experimental context and analysis pipeline.
Table 1: Overall Correlation Between RNA-seq and qPCR
| Measurement Type | Correlation Coefficient | Experimental Context | Source |
|---|---|---|---|
| Expression Correlation | R² = 0.798-0.845 (Pearson) | MAQC reference samples | [2] |
| Fold Change Correlation | R² = 0.927-0.934 (Pearson) | MAQCA vs. MAQCB samples | [2] |
| HLA Gene Expression | rho = 0.20-0.53 (Moderate) | HLA class I genes in PBMCs | [3] |
When examining differential expression, studies reveal that a significant majority of genes show consistent results between platforms. Analysis of the well-established MAQCA and MAQCB reference samples demonstrated that approximately 85% of genes showed consistent differential expression calls between RNA-seq and qPCR [2]. The remaining 15% represent genes with methodological discrepancies, though most of these (93%) showed relatively small differences in fold change (ΔFC < 2) [2].
Technology-Specific Performance Challenges
Both technologies exhibit specific limitations that can contribute to divergent results. For RNA-seq, alignment difficulties arise from reads that fail to map due to differences from reference genomes, particularly problematic for extremely polymorphic regions like HLA genes [3]. Additionally, cross-alignments between paralogs can artificially inflate expression estimates for certain gene families [3].
For qPCR, the method depends heavily on reference gene stability, with traditional housekeeping genes (e.g., ACTB, GAPDH) sometimes exhibiting unexpectedly high expression variance across biological conditions [7] [8]. One study found that previously established mosquito reference genes (RpL32, RpS17, ACT) were less stable than alternative candidates (eiF1A, eiF3j) in specific experimental conditions [8].
Table 2: Platform-Specific Limitations and Solutions
| Technology | Limitation | Impact | Mitigation Strategy |
|---|---|---|---|
| RNA-seq | Polymorphic regions | Underestimation of expression | HLA-tailored pipelines [3] |
| RNA-seq | Low expression genes | Reduced accuracy | Increased sequencing depth [79] |
| qPCR | Reference gene stability | Normalization errors | Genome-wide stability assessment [7] [8] |
| Both | Absolute quantification | Inaccurate measurements | Use relative quantification [79] |
Experimental Protocols for Method Comparison
Standardized RNA-seq Processing Workflows
Comprehensive benchmarking studies have evaluated multiple RNA-seq analysis workflows against whole-transcriptome qPCR data. The following methodology outlines a standardized approach for cross-platform validation:
Sample Preparation:
- Utilize well-characterized reference RNA samples (e.g., MAQCA Universal Human Reference RNA and MAQCB Human Brain Reference RNA) with built-in controls [2] [79].
- Include spike-ins of synthetic RNA from the External RNA Control Consortium (ERCC) to assess technical performance [79].
- Prepare mixture samples (3:1 and 1:3 ratios) to create samples with known expression relationships [79].
Library Preparation and Sequencing:
- Extract RNA using standardized kits (e.g., RNeasy Universal kit) with DNAse treatment to remove genomic DNA [3].
- For RNA-seq library preparation, employ either poly-A enrichment or rRNA depletion protocols depending on research goals.
- Sequence on multiple platforms (Illumina HiSeq, SOLiD, Roche 454) to assess platform-specific effects [79].
Bioinformatic Processing:
- Process reads through multiple workflows: Tophat-HTSeq, Tophat-Cufflinks, STAR-HTSeq, Kallisto, and Salmon [2].
- For challenging gene families (e.g., HLA genes), employ specialized pipelines that account for known diversity in the alignment step [3].
- Quantify expression using both gene-level and transcript-level approaches where applicable.
qPCR Validation Methodology
Assay Design:
- Design primers to detect specific transcripts that contribute proportionally to gene-level quantification [2].
- For comparative studies, align qPCR assays with transcripts considered for RNA-seq quantification to ensure equivalent targets [2].
Reference Gene Selection:
- Instead of relying solely on traditional housekeeping genes, employ RNA-seq data to identify stably expressed genes specific to the biological system [7] [8].
- Use coefficient of variation calculations or fold change cut-offs to assess expression stability across samples [7].
- Validate candidate reference genes using algorithms such as GeNorm, NormFinder, or BestKeeper [8].
Normalization and Analysis:
- Normalize qPCR data using stably expressed reference genes identified through RNA-seq analysis.
- Compare relative quantification values (ΔΔCq) with RNA-seq expression estimates (TPM, FPKM) for cross-platform correlation.
Figure 1: Experimental workflow for RNA-seq and qPCR comparison studies
Technological Frameworks and Analytical Pathways
Specialized RNA-seq Applications
For specific research applications, standard RNA-seq pipelines may require modification to address unique genomic challenges. The MORE-RNAseq pipeline was developed to quantify expression of retrotransposition-capable LINE-1 elements (rc-L1s), which are difficult to assess accurately due to their repetitive nature and complex genomic organization [81]. This approach involves:
- Manual curation of rc-L1 references to exclude repetitive sequence regions that cause erroneous mapping
- Creation of customized reference files that include both standard genes and curated rc-L1s
- Simultaneous quantification of gene and rc-L1 expression from standard RNA-seq data [81]
In the HLA system, specialized computational pipelines have been developed to address challenges posed by extreme polymorphism. These pipelines:
- Account for known HLA diversity during alignment rather than relying on a single reference genome
- Minimize cross-alignments between highly similar paralogs
- Have been shown to provide more accurate expression levels for HLA genes compared to standard approaches [3]
Reference Gene Selection Framework
The selection of appropriate reference genes for qPCR normalization can be systematically improved using RNA-seq data:
Transcriptome-Wide Screening:
- Calculate expression stability metrics (coefficient of variation, standard deviation) across all genes in the transcriptome
- Apply filtering criteria to identify genes with high, stable expression: TPM > 0 in all samples, standard variation of log2(TPM) < 1, average log2(TPM) > 5 [8]
- Use the coefficient of variation (< 0.2) to further refine candidate genes
Experimental Validation:
- Test top candidate genes using qPCR stability algorithms (GeNorm, NormFinder, BestKeeper)
- Select the most stable reference genes specific to the experimental conditions
- Avoid traditional housekeeping genes without stability verification [8]
Tools such as GSV (Gene Selector for Validation) automate this process, identifying both stable reference candidates and variable genes suitable for experimental validation [8].
Figure 2: Bioinformatics workflow for reference gene selection from RNA-seq data
Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Cross-Platform Validation
| Category | Product/Solution | Function | Application Notes |
|---|---|---|---|
| Reference Materials | MAQCA/B Reference RNA | Standardized expression profiling | Enables cross-platform comparison [2] [79] |
| Reference Materials | ERCC Spike-in Controls | Technical performance assessment | Evaluates detection limits and dynamic range [79] |
| RNA Extraction | RNeasy Universal Kit | High-quality RNA isolation | Includes DNAse treatment for genomic DNA removal [3] |
| qPCR Analysis | GeNorm/NormFinder/BestKeeper | Reference gene stability assessment | Statistical evaluation of candidate genes [8] |
| RNA-seq Analysis | HLA-tailored Pipelines | Specialized expression quantification | Addresses polymorphism challenges in HLA genes [3] |
| RNA-seq Analysis | MORE-RNAseq Pipeline | Retrotransposon expression quantification | Customized for LINE-1 elements [81] |
| Validation Tools | GSV Software | Reference candidate identification | Selects genes from RNA-seq data for qPCR validation [8] |
The concordance between RNA-seq and qPCR for gene expression analysis is generally strong, with approximately 85% of genes showing consistent differential expression calls between platforms [2]. However, methodological differences can lead to divergent results for specific gene classes, including highly polymorphic genes, low-expression transcripts, and members of complex gene families.
Researchers can optimize cross-platform validation through several key strategies: employing specialized computational pipelines for challenging genomic regions [3] [81], using RNA-seq data to identify stable reference genes for qPCR normalization [7] [8], and implementing standardized reference materials and spike-in controls to assess technical performance [2] [79]. As RNA-seq continues to evolve toward clinical applications, understanding the sources of methodological discordance will be essential for developing robust analytical frameworks that leverage the complementary strengths of both technologies.
Defining the 'When' and 'Why' of Orthogonal Validation by qPCR
Next-generation sequencing technologies, particularly RNA-Seq, have become the capstone method for genome-wide expression profiling, enabling the unbiased discovery of differentially expressed genes across the entire transcriptome [4] [24]. However, despite its power and prevalence, the question of whether RNA-Seq results require independent verification remains a common consideration in research and publication workflows. Orthogonal validation—the practice of verifying results using a method based on different biological or technical principles—provides a critical strategy for confirming key findings. Among the available methods, quantitative PCR (qPCR) has emerged as the most widely adopted technique for this purpose [82] [24]. This guide objectively examines the performance of qPCR as a validation tool for RNA-Seq data, providing researchers with evidence-based criteria for determining when such validation is necessary and how to implement it effectively within the context of differential expression research.
Technical Comparison: qPCR vs. RNA-Seq
Fundamental Methodological Differences
While both qPCR and RNA-Seq quantify gene expression, they operate on fundamentally different technical principles, which accounts for their complementary strengths. qPCR relies on the specific amplification of targeted cDNA sequences using predefined primers and fluorescent probes, providing extremely sensitive and precise quantification of a limited number of genes [82]. In contrast, RNA-Seq comprehensively sequences the entire transcriptome without prior target selection, enabling discovery but introducing different technical challenges related to library preparation, sequencing depth, and bioinformatic analysis [4] [3].
Performance Characteristics and Concordance
Table 1: Comparative Performance of qPCR and RNA-Seq
| Performance Metric | qPCR | RNA-Seq | Concordance Notes |
|---|---|---|---|
| Sensitivity | High (detects few copies) [82] | Variable (depends on sequencing depth) [4] | RNA-seq may miss low-abundance transcripts [4] |
| Dynamic Range | ~7-8 logs [83] | ~5 logs [4] | qPCR offers superior range for extreme expressions |
| Precision | High (low technical variation) [24] | Moderate (subject to more variables) [4] [3] | qPCR shows less technical noise |
| Multiplexing Capacity | Low (typically 1-6 targets per reaction) | Very High (entire transcriptome) | Complementary strengths |
| Throughput | Low to medium (focused targets) | High (all transcripts) | RNA-seq superior for discovery |
| Analytical Flexibility | Low (requires pre-defined targets) | High (post-hoc analysis) | RNA-seq allows re-analysis |
Evidence from large-scale comparisons reveals important insights about the agreement between these technologies. A comprehensive analysis by Everaert et al. compared five RNA-Seq analysis pipelines to wet-lab qPCR results for over 18,000 protein-coding genes [4]. The study found that 15-20% of genes showed non-concordant results (defined as differential expression in opposing directions, or one method showing differential expression while the other does not). However, the vast majority of these non-concordant cases (approximately 93%) exhibited fold changes lower than 2, with about 80% showing fold changes lower than 1.5 [4]. Importantly, only approximately 1.8% of genes showed severe non-concordance, and these were typically shorter genes expressed at low levels [4].
Special Considerations for Complex Genomic Regions
The extreme polymorphism and sequence similarity among paralogs within certain gene families present particular challenges for RNA-Seq quantification. Studies of Human Leukocyte Antigen (HLA) genes have demonstrated only moderate correlation (0.2 ≤ rho ≤ 0.53) between expression estimates from qPCR and RNA-Seq for HLA-A, -B, and -C genes [3]. This discrepancy arises from difficulties in aligning short reads to highly polymorphic regions and cross-alignments between similar paralogous sequences [3]. In such challenging genomic contexts, qPCR validation becomes particularly valuable due to its ability to target specific sequences with high specificity.
When is Orthogonal Validation by qPCR Necessary?
Research Scenarios Mandating Validation
Table 2: Decision Framework for qPCR Validation of RNA-Seq Results
| Scenario | Validation Recommended? | Rationale | Experimental Design Considerations |
|---|---|---|---|
| Small number of biological replicates | Yes [4] [24] | Limited statistical power increases false discovery risk | Use qPCR on additional samples to confirm biological effect |
| Low-fold change differences (<1.5-2) | Yes [4] | Most non-concordant results occur in this range | Ensure technical precision with sufficient qPCR replicates |
| Low-expression transcripts | Yes [4] | Higher technical variability in RNA-Seq quantification | Prioritize most biologically relevant low-expression targets |
| Complex genomic regions (e.g., HLA) | Yes [3] | RNA-Seq alignment challenges in polymorphic regions | Design allele-specific qPCR assays |
| Entire story relies on few genes | Yes [4] [24] | Ensures foundational results are robust | Validate all critical genes supporting main conclusions |
| RNA-Seq as preliminary screening | No [24] | Resources better spent on downstream functional studies | Focus validation on protein level or functional assays |
| Adequate replicates & high confidence results | Optional [4] [24] | Modern RNA-Seq pipelines are generally robust | Consider independent RNA-Seq cohort instead |
| Large-fold changes in well-expressed genes | Optional [4] | High concordance expected in these cases | Validation may still be requested for publication |
The decision to validate RNA-Seq results with qPCR should be guided by specific research circumstances and quality considerations. Dr. Christopher Mason from Weill Cornell Medicine emphasizes that in his research on cancer and space medicine, "whenever we see an interesting mutation, we validate it with an orthogonal assay like qPCR," which he considers "the high bar for validation" due to its high sensitivity [82].
Practical Workflow for Validation Decisions
Experimental Design for Orthogonal Validation
Best Practices in qPCR Experimental Design
Effective orthogonal validation requires careful experimental design. When designing qPCR assays to validate RNA-Seq results, several critical factors must be addressed:
Reference Gene Selection: Historically, housekeeping genes were assumed to have stable expression across tissues, but research has shown that even these genes can vary depending on tissue type, disease state, or stress conditions [82]. Proper validation requires using multiple reference genes (such as GAPDH or ribosomal genes) that have been verified for expression stability in the specific biological context under study [82] [7]. RNA-Seq data itself can be leveraged to identify stably expressed genes for use as references [7].
Sample Considerations: For the most robust validation, qPCR should be performed on an independent set of samples with proper biological replication, not just the same RNA used for RNA-Seq [24]. This approach validates both the technology and the underlying biological response.
Replication Strategy: Include both biological and technical replicates to account for different sources of variability [82]. Technical replicates assess assay precision, while biological replicates confirm consistent effects across different samples.
Implementing the Orthogonal Validation Principle
The orthogonal validation principle extends beyond simply confirming results with a different technology. As utilized in antibody validation, an orthogonal strategy involves "cross-referencing antibody-based results with data obtained using non-antibody-based methods" to identify "effects or artifacts that are directly related to" the primary method [84]. In the context of RNA-Seq and qPCR, this means recognizing that each method has distinct technical artifacts and biases, and consistency between them provides stronger evidence for biological reality than either method alone.
Research Reagent Solutions for Validation Experiments
Table 3: Essential Reagents and Materials for qPCR Validation Studies
| Reagent/Material | Function | Examples/Considerations |
|---|---|---|
| Nucleic Acid Isolation Kits | High-quality RNA extraction | AllPrep DNA/RNA Mini Kit (Qiagen) [31] |
| Reverse Transcription Kits | cDNA synthesis from RNA | Systems with high efficiency and minimal bias |
| qPCR Master Mixes | Amplification and detection | LightCycler 480 Probe Master (Roche) [83], QPCR Mastermix Plus (Eurogentec) [83] |
| Primer/Probe Sets | Target-specific detection | Allele-specific primers for patient-specific targets [83] |
| Reference Gene Assays | Normalization controls | Multiple genes (GAPDH, ribosomal proteins) [82] [7] |
| Quality Control Instruments | Assess nucleic acid quality | Qubit (quantitation), TapeStation (integrity) [31] |
| qPCR Instrumentation | Amplification and detection | LightCycler 480 (Roche), QuantStudio 5 (Thermo Fisher) [31] |
Orthogonal validation of RNA-Seq results by qPCR remains an important component of rigorous scientific research, particularly in contexts where findings will influence significant scientific conclusions or clinical applications. While modern RNA-Seq methods have substantially improved in reliability, judicious application of qPCR validation strengthens research outcomes in specific scenarios: when biological replication is limited, when fold changes are small, when studying challenging genomic regions, and when key conclusions rest on a small number of genes.
The evolving landscape of genomic technologies continues to refine our approach to validation. As noted by experts, "if all experimental steps and data analyses are carried out according to the state-of-the-art, results from RNA-seq are expected to be reliable" [4]. Nevertheless, qPCR maintains its position as a highly sensitive and reliable orthogonal method that can increase confidence in RNA-Seq findings, facilitate publication in peer-reviewed journals, and ultimately strengthen the foundation of scientific knowledge in genomics research.
The accurate quantification of gene expression is a cornerstone of modern genomics, influencing everything from basic research to clinical diagnostics. This challenge is particularly acute for extremely polymorphic gene families, such as the Human Leukocyte Antigen (HLA) genes, which play critical roles in immune function, disease susceptibility, and drug response [3]. These genes exhibit extensive sequence diversity between individuals and paralogs, creating substantial technical hurdles for both traditional and next-generation quantification methods. Within the broader thesis of RNA-seq versus qPCR validation in differential expression research, this case study examines the specific limitations and advantages of each platform when applied to complex gene families. The fundamental issue stems from the fact that standard RNA-seq analysis pipelines, which typically align short reads to a single reference genome, do not provide a complete representation of the vast allelic diversity present in polymorphic loci [3]. Consequently, reads originating from diverse alleles may fail to align properly or may align incorrectly to similar paralogous sequences, leading to biased expression estimates. This case study systematically compares the performance of qPCR and RNA-seq for expression quantification of polymorphic genes, evaluates specialized computational solutions, and provides a framework for researchers navigating these complex genomic regions.
Experimental Protocols: Benchmarking qPCR and RNA-seq for HLA Genes
Sample Preparation and Core Methodology
A direct comparative study was performed on a set of 96 healthy individuals for whom matched expression data were available using multiple techniques [3]. The experimental workflow was designed to minimize technical variation and enable a fair comparison between platforms.
- Biological Samples: RNA was extracted from freshly isolated peripheral blood mononuclear cells (PBMCs) from 96 healthy blood donors. This cell type is physiologically relevant for HLA gene expression studies.
- RNA Handling: Total RNA was extracted using the RNeasy Universal kit (Qiagen), treated with RNAse-free DNAse to remove genomic DNA contamination, and quantitated using an HT RNA Lab Chip (Caliper, Life Sciences) to ensure quality and consistency across samples [3].
- qPCR Protocol: Traditional quantitative PCR was performed using gene-specific assays for HLA class I genes (HLA-A, -B, and -C). This method relies on targeted amplification with primers designed for specific HLA loci.
- RNA-seq Protocol: A whole-transcriptome RNA-seq assay was conducted on the same set of 96 samples. To address the inherent biases of standard RNA-seq for HLA genes, an HLA-tailored bioinformatic pipeline was employed. This specialized pipeline accounts for known HLA diversity during the alignment step, minimizing the misalignment of reads that differ significantly from the reference genome [3].
- Cell Surface Expression Analysis: For a subset of individuals, antibody-based flow cytometry was used to quantify HLA-C protein expression on the cell surface, providing a third, orthogonal measurement for comparison [3].
Key Comparative Findings
The correlation between expression estimates from qPCR and the specialized RNA-seq pipeline was only moderate, with Spearman's correlation coefficients (rho) ranging from 0.2 to 0.53 for HLA-A, -B, and -C [3]. This moderate correlation underscores the significant technical and biological factors that differentiate these methods and complicate cross-platform comparisons. The study concluded that multiple factors must be accounted for when comparing quantifications from different techniques, including differences in the specific molecular phenotypes being measured (e.g., primer binding efficiency vs. read alignment success) and the technical nuances of each platform [3].
Performance Benchmarking: Quantitative Comparison of Expression Platforms
The following tables summarize key performance metrics and characteristics of qPCR and RNA-seq when applied to polymorphic gene families, synthesizing data from multiple studies.
Table 1: Direct Performance Comparison of qPCR and RNA-seq for Gene Expression Analysis
| Performance Metric | qPCR | Standard RNA-seq | HLA-Tailored RNA-seq |
|---|---|---|---|
| Correlation with Orthogonal Data | Considered benchmark for validation [2] | Moderate correlation with qPCR [2] | Moderate correlation with qPCR (rho: 0.2-0.53 for HLA) [3] |
| Dynamic Range | Wide, but can be limited by primer efficiency | Very wide | Very wide |
| Multiplexing Capability | Low (typically 1-4 targets per reaction) | Very High (entire transcriptome) | High (custom targeted panels or whole transcriptome) [85] |
| Ability to Detect Novel Alleles/Variants | No (requires pre-designed primers) | Yes, in theory | Yes, with appropriate tools |
| Throughput | Low to medium | High | High |
| Cost per Sample | Low for few targets, high for many | Moderate to High | Moderate to High (targeted can be lower) |
Table 2: Broader Benchmarking of RNA-seq Workflows Against qPCR Gold Standard
| RNA-seq Analysis Workflow | Expression Correlation with qPCR (R²) | Fold-Change Correlation with qPCR (R²) | Non-Concordant Genes on DE* |
|---|---|---|---|
| Salmon | 0.845 | 0.929 | 19.4% |
| Kallisto | 0.839 | 0.930 | 18.7% |
| Tophat-HTSeq | 0.827 | 0.934 | 15.1% |
| Tophat-Cufflinks | 0.798 | 0.927 | 17.8% |
| STAR-HTSeq | 0.821 | 0.933 | 15.3% |
Note: DE = Differential Expression. Non-concordant genes are those for which RNA-seq and qPCR disagree on differential expression status. Data adapted from a study on MAQCA/MAQCB reference samples [2].
Specialized Computational Methods for RNA-seq Analysis
The standard "align-then-count" RNA-seq workflow is often insufficient for polymorphic genes. In response, several computational strategies have been developed.
Methods for Bulk RNA-seq Data
For differential expression analysis from bulk RNA-seq, both read alignment-based and pseudoalignment-based methods are available. Their characteristics are summarized below.
Table 3: Selected Software Packages for Differential Expression Analysis from RNA-seq Data
| Method | Core Methodology | Read Count Distribution Assumption | Key Features / Normalization |
|---|---|---|---|
| edgeR | Empirical Bayes estimation and exact tests [86] | Negative Binomial | Robust for experiments with small numbers of replicates; uses TMM normalization [86] |
| DESeq | Similar to edgeR but models mean-variance relationship [86] | Negative Binomial | Balanced DE gene selection across dynamic range; uses DESeq sizeFactors [86] |
| limma | Transformation of counts for linear modeling [86] | After voom transformation, uses linear models | Empirical Bayes method on transformed data; can use TMM normalization [86] |
| Cuffdiff 2 | t-test on transcript-level estimates [86] | Beta Negative Binomial | Estimates expression at transcript level; can use geometric or quartile normalization [86] |
| NOIseq | Non-parametric method [86] | Non-parametric | Compares fold changes and absolute differences to a null distribution; uses RPKM/TMM/upper quartile [86] |
Methods for Single-Cell RNA-seq (scRNA-seq) Data
Single-cell data introduces additional challenges like multimodality and "drop-out" events (an abundance of zero counts), requiring even more specialized tools [87]. Methods like SCDE and MAST use two-part models to account for drop-outs, while scDD is designed to identify genes with different expression distributions across conditions, including changes in modality [87]. A comparative evaluation of eleven such tools revealed generally low agreement among them, highlighting that the choice of method significantly impacts research outcomes [87].
Visualizing Experimental and Analytical Workflows
The following diagram illustrates the core workflows for qPCR and RNA-seq quantification of polymorphic genes, highlighting the points of divergence that lead to technical challenges.
Successfully navigating expression analysis of polymorphic genes requires a carefully selected set of reagents and computational resources.
Table 4: Key Research Reagent Solutions for Quantifying Polymorphic Genes
| Reagent / Resource | Function / Application | Key Considerations |
|---|---|---|
| High-Quality RNA Extraction Kits (e.g., Qiagen RNeasy) | Isolation of intact, DNA-free total RNA from relevant tissues or cells. | Critical for all downstream assays; quality and integrity directly impact quantification accuracy [3]. |
| Locus-Specific qPCR Assays | Targeted quantification of specific genes (e.g., HLA-A) via amplification. | Primer design is critical; must target conserved regions of the locus to avoid allele-specific bias [3]. |
| Whole-Transcriptome Library Prep Kits | Preparation of sequencing libraries for RNA-seq from RNA samples. | Choice of kit can influence library complexity and coverage uniformity, affecting final counts. |
| HLA Allele Database & Genotyping Data | A comprehensive reference of known alleles for a polymorphic gene family. | Essential for specialized bioinformatic pipelines to accurately assign reads and quantify expression [3]. |
| Reference Samples with Orthogonal Data (e.g., MAQCA/MAQCB) | Benchmarked samples with expression data from multiple platforms (qPCR, microarray). | Allow for calibration and validation of new RNA-seq pipelines and methods [2]. |
| Specialized Bioinformatics Pipelines | Software tailored for aligning and quantifying reads from polymorphic loci. | Moves beyond a single reference genome to account for population diversity, reducing bias [3]. |
Quantifying the expression of extremely polymorphic gene families remains a formidable challenge in genomics. While qPCR provides a targeted and often more reliable benchmark, it lacks the scalability and discovery power of RNA-seq. Standard RNA-seq workflows, in turn, are prone to reference bias and misalignment when faced with high sequence diversity. The path forward lies in the continued development and adoption of specialized computational methods, such as HLA-tailored alignment pipelines, that explicitly account for genetic variation. Furthermore, the research community would benefit from standardized benchmarking using well-characterized reference materials. For researchers and drug development professionals, the key recommendation is to employ a tiered approach: using RNA-seq for discovery and broad profiling, followed by qPCR validation for critical polymorphic targets in physiologically relevant tissues. This integrated strategy, leveraging the strengths of both platforms while acknowledging their limitations, will yield the most robust and biologically meaningful results in the study of complex gene families.
Conclusion
RNA-seq and qPCR are not competing but deeply complementary technologies. RNA-seq excels as a powerful discovery tool for generating hypotheses, while qPCR remains indispensable for high-precision validation and targeted studies. The decision to validate RNA-seq findings with qPCR should be guided by the biological context: it is most critical when research conclusions hinge on a few genes, particularly those with low expression levels or small fold-changes. Future directions in biomedical research will be shaped by the development of more robust, standardized bioinformatics pipelines, the use of advanced reference materials for quality control, and the growing application of these integrated approaches in clinical diagnostics for detecting subtle, yet clinically relevant, differential expression.
References
- 1. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOorrHAzwMpO0SPNrqHQwHTDXE-Sv71OAK1ZIV85vddufnUZDzH6e]
- 2. Benchmarking of RNA-sequencing analysis workflows ... [https://www.nature.com/articles/s41598-017-01617-3]
- 3. Comparison between qPCR and RNA-seq reveals challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9883133/]
- 4. Do results obtained with RNA-sequencing require ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7823214/]
- 5. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 6. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 7. A Whole-Transcriptome Approach to Evaluating Reference ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5386857/]
- 8. RNA-seq validation: software for selection of reference and ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10511-y]
- 9. Real-Time PCR (qPCR) and Whole-Transcriptome NGS [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/ngs-comparison.html]
- 10. RNA-seq Data: Challenges in and Recommendations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4230301/]
- 11. Top Bioinformatics Tools for RNA Sequencing Data Analysis [https://riveraxe.com/top-bioinformatics-tools-for-rna-sequencing-data-analysis-best-value-for-your-research/]
- 12. Experimental validation of methods for differential gene ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-1767-y]
- 13. Powerful scRNA-seq Data Analysis Tools in 2025 [https://www.nygen.io/resources/blog/single-cell-scrna-seq-multi-omics-data-analysis-tools]
- 14. DNA Sequencing: How to Choose the Right Technology [https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/]
- 15. Next Generation Sequencing Platform: Ultimate Guide 2025 [https://lifebit.ai/blog/next-generation-sequencing-platform/]
- 16. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 17. A systematic benchmark of Nanopore long-read RNA ... [https://www.nature.com/articles/s41592-025-02623-4]
- 18. Comparison of direct cDNA and PCR-cDNA Nanopore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11507042/]
- 19. STALARD: Selective Target Amplification for Low-Abundance ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-025-01443-z]
- 20. From bench to bytes: a practical guide to RNA sequencing ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1697922/full]
- 21. Precision and Accuracy in Quantitative Measurement of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12603356/]
- 22. Gene Expression Analysis Market Outlook 2025-2030: [https://www.globenewswire.com/news-release/2025/05/21/3085487/0/en/Gene-Expression-Analysis-Market-Outlook-2025-2030-Overcoming-Challenges-such-as-Data-Complexities-and-Cost-Barriers.html]
- 23. Budgeting for an mRNA-seq project? How much does RNA ... [https://alitheagenomics.com/blog/budgeting-for-an-mrna-seq-project-here-are-the-main-cost-drivers-to-keep-an-eye-on]
- 24. Do I Need to Validate My RNA-Seq Results With qPCR? [https://www.rna-seqblog.com/do-i-need-to-validate-my-rna-seq-results-with-qpcr/]
- 25. Innovative qPCR Algorithm Using Platelet-Derived RNA for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11988175/]
- 26. The impact of PCR duplication on RNAseq data generated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12117910/]
- 27. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 28. RNA-seq data science: From raw data to effective ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.997383/full]
- 29. Real-Time PCR RNA Quantification Workflow [https://www.thermofisher.com/br/pt/home/life-science/pcr/real-time-pcr/real-time-pcr-applications/gene-expression-using-real-time-pcr/real-time-rna-quantification.html]
- 30. Impact of RNA-seq data analysis algorithms on gene ... [https://www.nature.com/articles/s41598-020-74567-y]
- 31. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 32. qPCR, Microarrays or RNA Sequencing - What to Choose? [https://biosistemika.com/blog/qpcr-microarrays-rna-sequencing-choose-one/]
- 33. a practical guide to RNA sequencing data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599993/]
- 34. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 35. A comparison of RNA-Seq data preprocessing pipelines for ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05801-x]
- 36. An updated comparison of microarray and RNA-seq for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11548-3]
- 37. Reference genes in real-time PCR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3825189/]
- 38. A comparison of normalization methods for the expression of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312509/]
- 39. Selecting reference genes in RT-qPCR based on ... [https://www.nature.com/articles/s41598-019-52217-2]
- 40. A stable combination of non-stable genes outperforms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11682138/]
- 41. Evaluation of normalisation strategies for qPCR data ... [https://www.nature.com/articles/s41598-025-06657-8]
- 42. Validated reference genes for normalization of RT-qPCR in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12222547/]
- 43. Evaluation and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-22650-7]
- 44. Selection of suitable reference genes for accurate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1557528/]
- 45. Selection and validation of RT-qPCR reference genes for multi ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12020-y]
- 46. Fold change based approach for identification of significant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8687202/]
- 47. Systematic comparison and assessment of RNA-seq ... [https://www.nature.com/articles/s41598-020-76881-x]
- 48. An evaluation of RNA-seq differential analysis methods [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0264246]
- 49. A comparison of methods for differential expression analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3608160/]
- 50. qPCR Troubleshooting: How to Ensure Successful ... [https://dispendix.com/blog/qpcr-troubleshooting]
- 51. The Ultimate qPCR Experiment: Producing Publication ... [https://www.sciencedirect.com/science/article/pii/S0167779918303421]
- 52. BLOG: Choosing the Right RNA Analysis Method [https://www.eurofins-viracorbiopharma.com/resource-library/posts/2025/june/blog-choosing-the-right-rna-analysis-method-rna-seq-nanostring-or-qpcr/]
- 53. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 54. Troubleshooting qPCR: Interpreting Amplification Curves [https://blog.biosearchtech.com/thebiosearchtechblog/bid/63622/qpcr-troubleshooting]
- 55. Experimental comparison of relative RT-qPCR quantification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2930637/]
- 56. What is a Ct Value and does it differ from qPCR Cq Values? [https://bitesizebio.com/24581/what-is-a-ct-value/]
- 57. High-Throughput Data Analysis for qPCR [https://www.neb.com/en-us/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr?srsltid=AfmBOoq3vKh1yzWty70GEbh_6zto_e_wgGUmLUO6OPkCnHfs982dEgEq]
- 58. Clinical validation of RNA sequencing for Mendelian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081282/]
- 59. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 60. Optimization Tips for Luna® qPCR [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/optimization-tips-for-luna-qpcr?srsltid=AfmBOoq7HfNvM0m7W5nGAE2-oswBHTJsMv7uX1x2TTk3qzRzBE3MxKKy]
- 61. Improving qPCR Efficiency: Primer Design and Reaction ... [https://synapse.patsnap.com/article/improving-qpcr-efficiency-primer-design-and-reaction-optimization]
- 62. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 63. PrimerQuest PCR & qPCR Primer Design Tool | IDT [https://www.idtdna.com/pages/tools/primerquest]
- 64. Real-time PCR (TaqMan) Primer and Probes Design Tool [https://www.genscript.com/tools/real-time-pcr-taqman-primer-design-tool]
- 65. Primer Optimization Using Temperature Gradient [https://www.merckmillipore.com/CR/es/technical-documents/protocol/genomics/qpcr/primer-optimization-using-temperature-gradient]
- 66. Pairwise efficiency : a new mathematical approach to qPCR ... data [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2911-5]
- 67. Bias in RNA-seq Library Preparation: Current Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8079181/]
- 68. NGS vs qPCR [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/ngs-vs-qpcr.html]
- 69. Introduction to RNA-Seq using high-performance computing [https://hbctraining.github.io/Intro-to-rnaseq-hpc-O2/lessons/04_alignment_quality.html]
- 70. Alignment - Bioinformatics for Beginners 2022 [https://bioinformatics.ccr.cancer.gov/docs/b4b/RNASeq_Overview/05.Alignment/]
- 71. CollectRnaSeqMetrics (Picard) - GATK - Broad Institute [https://gatk.broadinstitute.org/hc/en-us/articles/360037057492-CollectRnaSeqMetrics-Picard]
- 72. Variant calling from RNA-Seq data reveals allele-specific ... [https://www.nature.com/articles/s43856-025-00901-y]
- 73. iMapSplice: Alleviating reference bias through personalized ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0201554]
- 74. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 75. RNA-seq: An assessment of technical reproducibility and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2527709/]
- 76. Automation of RNA-Seq Sample Preparation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029635/]
- 77. Droplet-based high-throughput single microbe RNA ... [https://www.nature.com/articles/s41467-023-40137-9]
- 78. 10 Best Practices for Effective RNA-Seq Data Analysis - Focal [https://www.getfocal.co/post/10-best-practices-for-effective-rna-seq-data-analysis]
- 79. A comprehensive assessment of RNA-seq accuracy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4321899/]
- 80. Commentary: a review of technical considerations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522984/]
- 81. MORE-RNAseq: a pipeline for quantifying ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12138260/]
- 82. Cancer Research to Microbes: Harnessing the Power of ... [https://www.thermofisher.com/blog/behindthebench/harnessing-the-power-of-qpcr/]
- 83. Patient specific real-time PCR in precision medicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC9885152/]
- 84. Hallmarks of Antibody Validation: Orthogonal Strategy [https://www.cellsignal.com/about-us/approach-validation-principles/orthogonal-data?srsltid=AfmBOoqVOpinZNZ4Vffi7PK0DGD7FfRNrCym3sZpCnhOffKZ0nJ_ltFa]
- 85. Augmenting precision medicine via targeted RNA-Seq ... [https://www.nature.com/articles/s41698-025-00993-8]
- 86. Comparison of software packages for detecting differential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4293378/]
- 87. Comparative analysis of differential gene expression analysis ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2599-6]