Short-Read vs. Long-Read RNA Sequencing: A Comprehensive Guide for Biomedical Research and Drug Discovery
This article provides a definitive comparison of short-read and long-read RNA sequencing technologies for researchers and drug development professionals.
Short-Read vs. Long-Read RNA Sequencing: A Comprehensive Guide for Biomedical Research and Drug Discovery
Abstract
This article provides a definitive comparison of short-read and long-read RNA sequencing technologies for researchers and drug development professionals. It covers foundational principles, platform-specific methodologies, and application-specific guidance for tumor biology, single-cell analysis, and target discovery. The content addresses key challenges like cost-benefit optimization, sample quality, and data analysis, offering a clear framework for technology selection. By synthesizing validation data and emerging trends, this guide empowers strategic decision-making to leverage transcriptomics in advancing precision medicine and therapeutic development.
Core Technologies Demystified: How Short-Read and Long-Read Sequencing Work
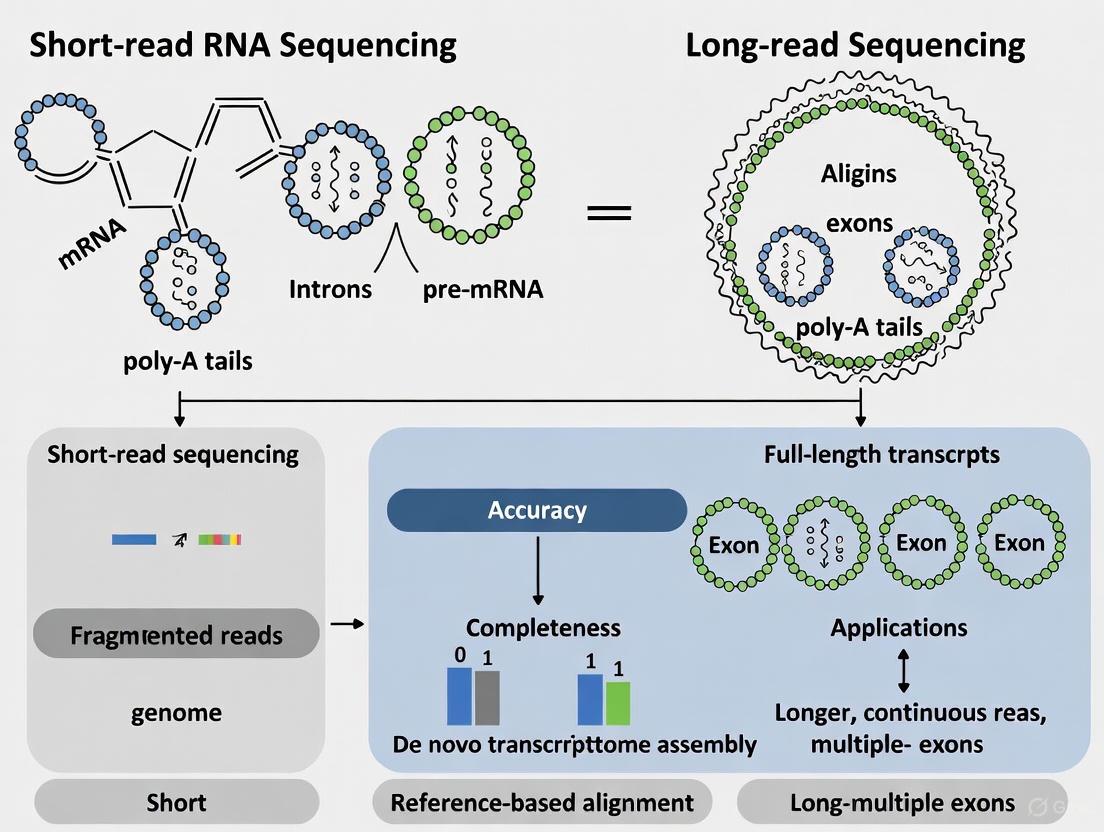
The foundational choice between short-read and long-read sequencing technologies profoundly shapes the design, outcome, and interpretation of RNA sequencing (RNA-seq) experiments. For over a decade, short-read sequencing (primarily Illumina) has been the undisputed gold standard for transcriptome profiling, offering high throughput and exceptional base accuracy [1]. Its dominance, however, is increasingly challenged by long-read sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), which enable the direct sequencing of full-length RNA transcripts in a single read [1] [2]. This capability is transformative for investigating the profound complexity of eukaryotic transcriptomes, where a single gene can produce numerous distinct isoforms through mechanisms like alternative splicing, alternative transcriptional start sites, and alternative polyadenylation [1]. While short-read methods infer this complexity indirectly by piecing together fragmented sequences, long-read technologies capture it directly, preserving the connectivity of distant exons [1]. This guide provides an objective comparison of these technologies, focusing on their core characteristics—read length, throughput, and chemistry—and summarizes key experimental data to inform researchers and drug development professionals navigating this evolving landscape.
Core Technical Specifications and Performance Comparison
The fundamental differences between short-read and long-read technologies are rooted in their underlying biochemistry and physics, leading to distinct performance profiles.
Table 1: Core Technical Specifications of Major RNA Sequencing Platforms
| Feature | Illumina Short-Read RNA-seq | PacBio Long-Read RNA-seq | ONT Long-Read RNA-seq |
|---|---|---|---|
| Typical Read Length | 50-300 bp [1] | Up to 25 kb [1] | Up to 4 Mb [1]; often 1,000-20,000+ bp [3] |
| Base Accuracy | ~99.9% [1] | ~99.9% (HiFi mode) [1] [3] | 95% - 99% (varies with chemistry) [1] |
| Throughput (per run/cell) | High (e.g., ~300,000 reads/cell in a scRNA-seq study [4]) | Moderate (improved with Kinnex/MAS-ISO-seq) [4] [1] | High (up to 277 Gb on PromethION flow cell) [1] |
| Core Chemistry | Sequencing-by-synthesis with fluorescently labelled nucleotides [5] | Single Molecule, Real-Time (SMRT) sequencing in zero-mode waveguides (ZMWs) [3] | Nanopore-based detection of ionic current changes [1] [5] |
| Key RNA-seq Applications | High-quality gene-level expression quantification [4] [6] | Full-length isoform discovery and quantification, variant detection [1] [3] | Full-length isoform analysis, direct RNA sequencing, detection of RNA modifications [1] [6] |
Short-read technology, exemplified by Illumina, is an ensemble method. It requires DNA polymerase and fluorescently labelled nucleotides to sequence millions of DNA clusters in parallel on a flow cell through sequencing-by-synthesis [5]. While it provides high-depth, high-accuracy data ideal for quantifying gene expression levels, its fundamental limitation is read length. The need to fragment transcripts before sequencing means the connectivity between distant exons is lost, making it challenging to resolve specific transcript isoforms [1].
In contrast, long-read platforms sequence single molecules. PacBio's HiFi sequencing employs circular consensus sequencing (CCS). DNA is circularized and sequenced multiple times by a polymerase immobilized at the bottom of a nanophotonic structure called a zero-mode waveguide (ZMW). This multi-pass approach generates a highly accurate consensus sequence (HiFi read) [3]. Oxford Nanopore's technology is physically distinct: it measures disruptions in an ionic current as a single RNA or DNA molecule is threaded through a protein nanopore. This allows for direct RNA sequencing without cDNA synthesis and enables the detection of RNA modifications [1] [6]. A key differentiator is that long reads can encompass a complete RNA transcript, directly revealing its full sequence and structure [2].
Experimental Comparisons and Benchmarking Data
Recent controlled studies provide empirical data on how these technical differences translate into practical performance.
Table 2: Key Findings from Comparative RNA-seq Studies
| Study (Source) | Experimental Design | Key Findings on Performance |
|---|---|---|
| Clear Cell Renal Cell Carcinoma (ccRCC) Organoids [4] | Same 10x Genomics 3' cDNA from patient-derived organoids sequenced on Illumina (NovaSeq) and PacBio (Sequel IIe). | - Short-reads: Higher sequencing depth, recovered more UMIs per cell.- Long-reads: Retained transcripts <500 bp, enabled removal of truncated cDNA artefacts. Data from both methods were "highly comparable" for gene expression. |
| Singapore Nanopore Expression (SG-NEx) Project [6] | Systematic benchmark of 5 protocols (Illumina, ONT direct RNA, ONT direct cDNA, ONT PCR-cDNA, PacBio IsoSeq) across 7 human cell lines. | - Throughput: PCR-amplified cDNA (ONT & Illumina) generated highest throughput.- Read Length: PacBio IsoSeq and ONT direct RNA produced the longest reads.- Coverage: Long-read protocols showed more uniform 5'/3' coverage; short-reads had more reads assigned to multiple transcripts.- Bias: PacBio IsoSeq was depleted of shorter transcripts; PCR-based protocols over-amplified highly expressed genes. |
| Colorectal Cancer Genomics [7] | Comparison of Illumina whole-exome and Nanopore whole-genome sequencing on patient samples. | - Coverage: Illumina provided higher depth over target regions (e.g., ~105X vs ~21X for cancer samples).- Mapping Quality: Both were >99% accurate, with Illumina slightly higher (99.96% vs 99.89%). |
The SG-NEx project, a comprehensive benchmarking effort, found that while gene expression estimates are robustly correlated across all major RNA-seq protocols, each method introduces distinct biases [6]. For instance, PCR-amplified protocols (common in both short-read and some long-read workflows) can over-represent the most highly expressed genes, while PacBio's IsoSeq protocol was found to be significantly depleted of shorter transcripts [6]. This highlights that the library preparation method, not just the sequencing technology itself, is a critical source of bias.
In single-cell RNA-seq, a direct per-molecule comparison found that both Illumina and PacBio methods recover a large proportion of cells and transcripts from the same cDNA library, rendering "highly comparable results" for relevant gene signatures [4]. However, platform-specific processing allowed long-read sequencing to filter out artefacts identifiable only from full-length transcript data, demonstrating a unique advantage in data quality control [4].
Core Chemistry and Workflow Visualization
The experimental workflows for short-read and long-read sequencing are fundamentally different, from library preparation to base detection.
Diagram 1: Core Chemistry of Major Sequencing Platforms
This diagram illustrates the fundamental biochemical processes underlying the three major sequencing platforms.
Experimental Workflow for a Comparative Study
A typical experimental design for directly comparing sequencing technologies, as performed in the ccRCC organoid study [4], involves several key stages.
This workflow visualizes the methodology for a direct, per-molecule comparison of short and long-read sequencing from the same cDNA library [4].
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of a comparative RNA-seq study requires careful selection of reagents and kits. The following table details key solutions used in the featured experiments.
Table 3: Key Research Reagent Solutions for RNA-seq Studies
| Item | Function | Example from Literature |
|---|---|---|
| 10x Genomics Chromium Single Cell 3' Kit | Partitions single cells into GEMs for barcoding and reverse transcription of full-length cDNA. | Used to generate the input cDNA for cross-platform sequencing in the ccRCC organoid study [4]. |
| PacBio MAS-ISO-seq for 10x Genomics Kit | Prepares 10x Genomics cDNA for long-read sequencing by removing TSO artefacts and concatenating transcripts. | Enabled high-throughput long-read scRNA-seq on the PacBio platform [4]. |
| Spike-in RNA Controls | Synthetic RNA molecules with known sequences and concentrations used to benchmark accuracy and quantification. | The SG-NEx project used Sequins, ERCC, and SIRVs to evaluate protocol performance [6]. |
| Solid-Phase Reversible Immobilization (SPRI) Beads | Used for post-reaction clean-up and size selection of cDNA libraries. | A standard step in both Illumina and PacBio library preparation protocols [4]. |
| Single-Molecule Real-Time (SMRT) Cell | The nanofluidic device containing millions of ZMWs where PacBio sequencing occurs. | The core consumable for PacBio sequencing runs [3]. |
| Nanopore Flow Cell (e.g., PromethION) | The device containing the nanopore array where ONT sequencing occurs. | The core consumable for ONT sequencing runs [1]. |
The choice between short-read and long-read RNA sequencing is not a simple matter of one technology being superior to the other. Instead, they offer complementary strengths. Short-read sequencing remains a powerful, cost-effective tool for applications where high-throughput, accurate gene-level quantification is the primary goal, such as differential gene expression studies in large cohorts [4] [6]. Long-read sequencing is transformative for applications that require resolving transcript isoform diversity, detecting fusion genes, characterizing non-coding RNAs, and identifying RNA modifications [1] [2]. Empirical data shows that while gene-level results are often highly correlated, long-reads provide a unique and often more accurate view of transcript-level biology [6].
The field continues to evolve rapidly. PacBio's Kinnex (formerly MAS-ISO-seq) and ONT's progressively more accurate chemistries are systematically addressing historical limitations of long-read technology, such as throughput and per-base accuracy [4] [1]. Concurrently, sophisticated computational tools and standardized pipelines like nf-core/nanoseq are maturing, making the analysis of long-read data more accessible [6]. For researchers and drug developers, the decision must be driven by the specific biological question. If the objective is to understand not just which genes are expressed but how they are spliced and processed into functional molecules, long-read RNA sequencing is increasingly becoming an indispensable, foundational technology [1] [8].
Short-read sequencing technologies are foundational to modern genomics, enabling high-throughput genetic analysis that drives research and drug development. These methods can be broadly categorized into three core biochemical approaches: Sequencing by Synthesis (SBS), Sequencing by Binding (SBB), and Sequencing by Ligation (SBL). Each technology employs distinct mechanisms for parallel sequencing of billions of DNA fragments, typically generating reads of 50 to 300 bases [9]. This guide provides an objective, data-driven comparison of these methodologies, detailing their operational principles, performance characteristics, and experimental considerations to inform scientific and clinical application choices.
Core Technologies and Methodologies
Sequencing by Synthesis (SBS)
SBS methods utilize DNA polymerase to synthesize a complementary strand to the DNA template. Nucleotide incorporation is detected via one of two primary methods:
- Fluorescently-Labeled Nucleotides with Reversible Blockers: The process involves the incorporation of a fluorescently-labeled nucleotide, which also contains a reversible terminator that halts the synthesis reaction. After imaging to identify the incorporated base, the fluorescent dye and blocker are chemically removed, allowing the next nucleotide to be incorporated [9]. This cyclical process is characteristic of platforms like Illumina.
- Unmodified Nucleotides with Sequential Addition: In this "sequencing-by-synthesis-by-pH-change" method, unmodified nucleotides (A, T, G, C) are flowed sequentially. The incorporation of a nucleotide by polymerase releases a hydrogen ion, causing a detectable local pH change. The signal is proportional to the number of identical nucleotides incorporated consecutively. Unincorporated nucleotides are washed away before introducing the next type [9] [10]. This principle is used by Ion Torrent technology.
Sequencing by Binding (SBB)
SBB also uses a polymerase enzyme but separates the nucleotide identification and incorporation steps, creating a more natural DNA synthesis process [10]. The workflow for a single base extension is as follows:
- A primer hybridized to the template DNA has a reversible blocker attached.
- Fluorescently-labeled nucleotides are introduced. The complementary nucleotide binds transiently to the template, and its fluorescent signal is imaged.
- Because of the blocker, the labeled nucleotide cannot be incorporated and is washed away.
- The blocker on the primer is then chemically removed, and unlabeled nucleotides with reversible blockers are added, allowing the polymerase to extend the DNA strand by a single base [9].
This technology is implemented in platforms like the Element Biosciences AVITI System [10].
Sequencing by Ligation (SBL)
SBL employs DNA ligase instead of polymerase to determine the sequence. The process uses short oligonucleotide probes of known sequence that are fluorescently labeled. The ligase enzyme preferentially joins the probe that perfectly matches the template strand. The fluorescent signal of the successfully ligated probe identifies the base sequence. After imaging, the complex is cleaved to remove the fluorescent label and prepare for the next ligation cycle [9]. A historical example of this technology is SOLiD sequencing, which is noted to struggle with palindromic sequences that can form hairpin structures and evade ligation [9] [10].
The following diagram illustrates the core logical workflow and key differences between these three primary short-read sequencing methods.
Performance Comparison and Experimental Data
The different chemistries of SBS, SBB, and SBL lead to distinct performance profiles, which are critical for experimental planning. The table below summarizes key quantitative and qualitative characteristics based on current technologies and literature.
Table 1: Comparative Performance of Short-Read Sequencing Technologies
| Feature | Sequencing by Synthesis (SBS) | Sequencing by Binding (SBB) | Sequencing by Ligation (SBL) |
|---|---|---|---|
| Read Length | 50-300 bp [9] | Up to 300 bp (e.g., AVITI System) [10] | 50-100 bp (historical) [10] |
| Primary Detection Method | Fluorescence (Illumina) or pH change (Ion Torrent) [9] [10] | Fluorescence (transient binding) [9] [10] | Fluorescence (ligation) [9] |
| Typical Accuracy | High (Q30+ common) [10] | Very High (Q40+ reported) [10] | High, but challenged by palindromes [9] |
| Throughput | Very High | High | Moderate to High (historical) |
| Library Prep Time | Varies; can be multistep [10] | Not specified in results | Multistep and laborious [10] |
| Key Strengths | High throughput, established workflows, low cost per base [11] [9] | High accuracy, reduced enzyme bias [10] | Robustness in some sequence contexts |
| Key Limitations | Amplification biases, short reads struggle with repeats [10] | Newer platform, smaller ecosystem | Inefficient with hairpin-forming sequences [9] |
| Example Platforms | Illumina, Ion Torrent [10] | Element Biosciences AVITI [10] | SOLiD (discontinued) [10] |
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of short-read sequencing requires a suite of specialized reagents and kits. The following table details key components used in typical workflows.
Table 2: Essential Research Reagent Solutions for Short-Read Sequencing
| Item | Function | Example Use Case |
|---|---|---|
| Library Preparation Kits | Fragment DNA, repair ends, add platform-specific adapters, and amplify the library. | Used in all short-read protocols to convert raw nucleic acids into a sequencer-compatible format [10]. |
| Platform-Specific Flow Cells/ Chips | Solid surface where clonal amplification and the sequencing reaction occur. | Illumina's patterned flow cells for bridge amplification; Ion Torrent's chips for pH detection [10] [12]. |
| Polymerase or Ligase Enzymes | Key enzyme driving the sequencing reaction (SBS/SBB: polymerase; SBL: ligase). | Highly engineered enzymes are critical for incorporating nucleotides (SBS) or binding probes (SBB) with high fidelity and efficiency [9]. |
| Fluorescently-Labeled Nucleotides/Probes | Identify the base sequence during the detection phase of the cycle. | Reversible terminators in Illumina SBS; fluorescent probes in SBL [9]. |
| Unique Dual Indexes (UDIs) | Barcode sequences added during library prep to multiplex samples. | Allows pooling and simultaneous sequencing of dozens of samples, reducing cost per sample [4]. |
| Solid-Phase Reversible Immobilization (SPRI) Beads | Magnetic beads for size selection and cleanup of DNA fragments between library prep steps. | Used for purifying and selecting appropriately sized cDNA libraries after amplification [4]. |
Contextualizing Short-Reads in the Broader Sequencing Landscape
While powerful, short-read technologies have inherent limitations. Their primary challenge is the inability to sequence long, continuous stretches of DNA. Genomes must be fragmented, and computer programs assemble these short reads into a continuous sequence. This process can fail in complex regions, leading to gaps and ambiguities, particularly in areas with large structural variations, highly repetitive sequences, or to resolve specific transcript isoforms [10] [6].
This limitation is the driving force behind the development and adoption of long-read sequencing technologies (PacBio HiFi and Oxford Nanopore). Long-reads can span entire repetitive elements or genes in a single read, simplifying genome assembly and enabling the direct detection of isoform-level expression in transcriptomics [13] [10]. However, long-read sequencing has historically faced challenges with higher error rates and cost, though these have improved dramatically [13] [10].
The choice between short-read and long-read technologies is therefore application-dependent. Short-reads remain the gold standard for high-throughput, cost-effective applications like variant calling, gene expression quantification (gene-level), and targeted sequencing [9]. In contrast, long-reads are indispensable for de novo genome assembly, resolving structural variants, and full-length transcript isoform analysis [13] [6].
The transition from short-read to long-read RNA sequencing represents a paradigm shift in transcriptomics. While conventional short-read methods (50-300 bases) have provided valuable gene-level expression data, their inherent limitations in resolving complex isoforms, alternative splicing events, and base modifications have constrained our understanding of transcriptional regulation [13] [8]. Long-read technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) now enable end-to-end sequencing of full-length transcripts, capturing the complete complexity of RNA molecules without the need for assembly [14]. This technological advancement is particularly crucial for researchers and drug development professionals investigating diseases where alternative splicing, novel isoforms, and RNA modifications play critical roles, such as in cancer, neurological disorders, and rare genetic conditions [15] [16].
The fundamental distinction between these platforms lies in their underlying chemistry and data output characteristics. PacBio's HiFi (High Fidelity) sequencing employs circular consensus sequencing (CCS) to generate highly accurate long reads (15-20 kb) with quality scores exceeding Q30 (99.9% accuracy) [13] [14]. In contrast, Oxford Nanopore Technologies sequences native RNA or DNA molecules by detecting changes in electrical current as nucleic acids pass through protein nanopores, enabling ultra-long reads (sometimes exceeding 100 kb) and direct detection of RNA modifications [13] [17]. Each approach offers distinct advantages for specific research applications, from comprehensive isoform characterization to real-time detection of epigenetic modifications.
Technology Comparison: PacBio HiFi vs. Oxford Nanopore
Core Methodologies and Performance Characteristics
The following table summarizes the fundamental technical specifications and performance metrics of both platforms, providing researchers with objective data for platform selection.
Table 1: Technical comparison of PacBio HiFi and Oxford Nanopore sequencing platforms
| Parameter | PacBio HiFi Sequencing | Oxford Nanopore Technologies |
|---|---|---|
| Technology Principle | Fluorescent detection of nucleotide incorporation by polymerase in SMRT cells | Measurement of current changes as molecules pass through protein nanopores |
| Read Length | 500 bp - 20 kb [13] | 20 kb to >4 Mb; can exceed 100 kb [13] |
| Raw Read Accuracy | ~99.9% (Q30+) [13] [14] | ~99% (Q20) with recent improvements [13] [18] |
| Typical Run Time | 24 hours [13] | Up to 72 hours [13] |
| Typical Yield per Flow Cell | 60-120 Gb [13] | 50-100 Gb [13] |
| Input Requirements | DNA, cDNA [13] | Native DNA, RNA, cDNA [13] [17] |
| DNA Modification Detection | 5mC, 6mA without bisulfite treatment [13] | 5mC, 5hmC, 6mA; direct detection [13] |
| Variant Calling | SNVs, indels, structural variants [13] | SNVs, structural variants; challenges with indels in repetitive regions [13] |
| Base Calling | On-instrument (no additional cost) [13] | Off-instrument, often requires costly GPU servers [13] |
| Portable Sequencing | Not available | MinION, Flongle available [13] [14] |
| File Storage Requirements | 30-60 GB (BAM format) [13] | ~1,300 GB (FAST5/POD5 format) [13] |
Workflow and Data Analysis Considerations
Beyond the technical specifications, practical implementation factors significantly impact platform selection. PacBio systems perform basecalling on-instrument, generating analysis-ready BAM files with minimal computational overhead [13]. In contrast, Oxford Nanopore requires substantial computational resources for basecalling, often necessitating expensive GPU servers that increase the total cost of ownership [13]. Storage requirements also differ dramatically, with Nanopore datasets (~1,300 GB per genome) demanding approximately 20 times more storage than PacBio outputs (30-60 GB per genome) [13].
For transcriptomics, both platforms offer distinct approaches. PacBio's HiFi sequencing of cDNA provides exceptional accuracy for isoform quantification and discovery, while Oxford Nanopore enables direct RNA sequencing that preserves native modification information [6] [17]. The selection between these approaches depends on the research priorities: accurate quantification of known and novel isoforms (PacBio) versus detection of RNA modifications alongside sequence information (ONT Direct RNA Sequencing).
Diagram 1: Technology selection workflow for long-read RNA sequencing
Direct RNA Sequencing: A Specialized Nanopore Application
Oxford Nanopore's Direct RNA Sequencing (DRS) represents a distinctive approach that sequences native RNA molecules without reverse transcription or amplification [17]. This methodology preserves base modifications and eliminates amplification biases, providing a direct view of the epitranscriptome. The workflow begins with RNA extraction followed by adapter ligation to the 3' poly(A) tail. The prepared library is then loaded onto flow cells where motor proteins unwind RNA molecules and guide them through nanopores. As each RNA molecule passes through the pore, distinct current disruptions corresponding to specific RNA bases and their modifications are recorded in real-time [17].
Recent advancements in Nanopore chemistry, particularly the RNA004 kit with updated motor proteins and 9-mer signal detection, have substantially improved basecalling accuracy compared to previous versions [19] [17]. However, DRS still faces challenges with complete 5' end coverage since sequencing initiates at the 3' poly(A) tail, potentially missing information about 5' cap structures and beginning of transcripts [6]. Despite this limitation, the ability to simultaneously detect sequence information and RNA modifications in a single assay makes DRS uniquely valuable for studying the functional role of epitranscriptomic modifications in development, disease, and therapeutic response [19].
Experimental Design Considerations
Effective Direct RNA Sequencing requires careful experimental planning. The recommended input is 500 ng of poly(A)-enriched RNA, though lower inputs can be accommodated with potential trade-offs in library complexity [17]. Unlike cDNA-based approaches, DRS does not require fragmentation or amplification, simplifying library preparation but potentially introducing biases based on RNA secondary structure and modification density. Researchers should include appropriate controls, such as in vitro transcribed (IVT) RNA, to distinguish true modifications from sequence-specific artifacts [19].
The bioinformatic analysis of DRS data demands specialized tools for basecalling, alignment, and modification detection. The standard workflow includes raw signal processing with Guppy or Dorado basecallers, alignment with minimap2 or GraphMap, and modification detection with specialized tools like m6Anet or Nanocompore [19] [17]. Computational requirements remain substantial, with basecalling typically requiring GPU acceleration and significant storage capacity for raw signal data (FAST5/POD5 files).
Diagram 2: Nanopore Direct RNA Sequencing workflow and advantages
Performance Benchmarking and Experimental Evidence
Transcriptomics Applications
Recent comprehensive benchmarking studies provide critical insights into platform performance for transcript-level analysis. The Singapore Nanopore Expression (SG-NEx) project compared five RNA-seq protocols across seven human cell lines, offering one of the most systematic comparisons to date [6]. This study found that PacBio IsoSeq generated the longest reads on average and, together with Nanopore's PCR-amplified cDNA protocol, showed the most uniform coverage across transcript lengths and the highest proportion of reads spanning all exon junctions ("full-splice-match reads") [6].
For gene expression quantification, Nanopore long-read RNA-seq demonstrated the lowest estimation error and highest correlation with known spike-in RNA concentrations across multiple computational quantification methods [6]. However, PacBio's HiFi sequencing consistently outperforms for variant detection, with one study showing it detected approximately three times more true positive single nucleotide variants (SNVs) than Oxford Nanopore, making it particularly valuable for allele-specific expression studies [16]. The exceptional accuracy of HiFi reads also enables reliable detection of insertions and deletions (indels), which remains challenging for Nanopore technology, particularly in repetitive regions [13].
Table 2: Performance comparison in recent benchmarking studies
| Application | PacBio HiFi Performance | Oxford Nanopore Performance | Reference Study |
|---|---|---|---|
| Full-length Transcript Detection | Identified >180,000 mRNA isoforms (>50% novel) in lung adenocarcinoma [15] | Robust identification of major isoforms; lower uniformity with direct RNA [6] | SG-NEx [6] |
| SNV Detection | ~3× more true positives compared to ONT [16] | Lower SNP calling performance due to higher error rates [16] | HPRC Kinnex [16] |
| Species-level Taxonomic Resolution | 63% of sequences classified to species level [18] | 76% of sequences classified to species level [18] | Rabbit gut microbiota [18] |
| RNA Modification Detection | Not applicable for direct RNA modification detection | m6A detection: Dorado recall ~0.92, m6Anet recall ~0.51 at ≥10% modification sites [19] | RNA004 benchmarking [19] |
| Differential Expression Analysis | Strong concordance with Illumina (Pearson >0.9 gene level) with lower inferential variability [16] | High correlation with expected spike-in concentrations; some protocol-specific biases [6] | Kinnex benchmarking [16] |
Specialized Research Applications
Ultra-low Input Sequencing
Recent advancements have extended long-read sequencing to challenging sample types. PacBio's ultralow-input (ULI) protocol, now refined as the AmpliFi protocol, enables comprehensive variant detection with as little as 1-10 ng of input DNA [15]. This capability is particularly valuable for clinical samples where material is limited, such as tumor biopsies, fine-needle aspirates, and single cells. In application to hereditary colorectal cancer samples, ULI-HiFi sequencing revealed progressive tandem repeat expansion in a tumor suppressor gene across normal tissue, polyp, and adenocarcinoma samples, demonstrating the power of long-read sequencing for capturing dynamic genomic changes in disease progression [15].
Epigenetics and Methylation Profiling
For epigenomic studies, PacBio HiFi sequencing provides a more complete view of the DNA methylome compared to whole-genome bisulfite sequencing (WGBS). In a twin study, HiFi sequencing identified approximately 5.6 million more CpG sites than WGBS, particularly in repetitive elements and regions of low coverage with bisulfite-based methods [15]. The coverage pattern of HiFi sequencing showed a uniform distribution peaking at 28-30×, with over 90% of CpGs achieving ≥10× coverage, compared to approximately 65% in WGBS datasets [15]. This comprehensive coverage enables de novo DNA methylation analysis, reporting CpG sites beyond reference sequences without the DNA damage associated with bisulfite conversion.
Repeat Expansion Disorders
Long-read sequencing has revolutionized the diagnosis of repeat expansion disorders that often evade detection by short-read technologies. In one study of Familial Adult Myoclonic Epilepsy type 3 (FAME3), PacBio HiFi sequencing identified a pathogenic MARCHF6 intronic expansion that had been missed by multiple rounds of exome and genome testing [15]. The analysis revealed that affected individuals carried one allele with 15 TTTTA repeats and a second allele with a compound expansion of 661 TTTTA and 12 TTTCA repeats, with increasing repeat sizes in later generations [15]. This study highlighted that disease manifestation requires TTTCA repeats in tandem with TTTTA motifs, demonstrating the importance of assessing both repeat length and composition—a capability uniquely provided by long-read sequencing.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key research reagents and computational tools for long-read RNA sequencing
| Item | Function | Example Products/Platforms |
|---|---|---|
| Library Preparation Kits | Convert RNA to sequence-ready libraries | PacBio Kinnex RNA Single-Cell Kit, ONT Direct RNA Sequencing Kit (SQK-RNA004) |
| Polymerase Enzymes | Amplify cDNA for sequencing | KAPA HiFi HotStart ReadyMix (PacBio), Long Amp Taq (Nanopore) |
| Barcoding Systems | Multiplex samples in a single run | PacBio Multiplexed Barcoded Adapters, ONT Native Barcoding kits |
| Flow Cells/Consumables | Platform-specific sequencing substrates | SMRT Cells (PacBio), MinION/PromethION Flow Cells (ONT) |
| Basecalling Software | Convert raw signals to nucleotide sequences | Dorado (ONT), SMRT Link (PacBio) |
| Modification Detection Tools | Identify RNA modifications from sequencing data | m6Anet, Nanocompore (ONT) |
| Alignment & Quantification | Map reads and quantify expression | Minimap2, StringTie, Bambu |
| Quality Control Tools | Assess read quality and library preparation | NanoPlot (ONT), SMRT Link Quality Control (PacBio) |
| Reference Databases | Taxonomic classification and annotation | SILVA, Greengenes (16S rRNA); GENCODE, RefSeq (mRNA) |
The choice between PacBio HiFi and Oxford Nanopore technologies depends fundamentally on research priorities. PacBio's exceptional accuracy (Q30+) makes it ideally suited for applications requiring high-confidence variant calling, including SNVs, indels, and structural variants [13] [16]. This precision is particularly valuable in clinical research and diagnostic development where false positives carry significant consequences. Additionally, PacBio's uniform coverage and lower computational requirements provide practical advantages for laboratories with limited bioinformatics infrastructure [13].
Oxford Nanopore offers distinctive capabilities through its Direct RNA Sequencing platform, enabling simultaneous detection of sequence information and RNA modifications without additional chemical treatments or conversion steps [19] [17]. The platform's portability and real-time sequencing capabilities further expand its utility for field applications and rapid diagnostics [13] [14]. However, these advantages come with higher computational demands for basecalling and substantially larger storage requirements for raw signal data [13].
For drug development professionals, these technologies open new avenues for biomarker discovery, therapeutic target identification, and understanding drug mechanisms at the transcriptome level. The ability to fully characterize isoform-specific expression, allele-specific regulation, and epitranscriptomic modifications provides unprecedented insight into disease mechanisms and treatment responses [15] [16]. As these technologies continue to evolve, with both platforms demonstrating rapid improvements in accuracy, throughput, and accessibility, long-read RNA sequencing is positioned to become a foundational technology for both basic research and translational applications.
In the field of genomics, the fundamental requirement for nearly all applications is accurate base calling. The inherent limitations of sequencing technologies, however, introduce errors that researchers must carefully manage. This challenge is particularly pronounced in long-read sequencing, which, despite providing invaluable long-range genomic information, has historically been hampered by higher error rates compared to short-read technologies [1]. To bridge this accuracy gap, sophisticated computational methods have been developed, with circular consensus sequencing (CCS) emerging as a powerful approach for generating highly accurate long reads [20].
This guide provides a objective comparison of the accuracy and error profiles of modern sequencing platforms, focusing on the critical role of quality scores (Q scores) and consensus methods. We present summarized experimental data, detailed protocols, and analytical tools to help researchers and drug development professionals navigate the evolving landscape of sequencing technologies for their RNA research.
Understanding Q Scores and Consensus Sequencing
The Metric of Accuracy: Q Scores
In sequencing data, a Q score (or Phred quality score) is a logarithmic measurement that predicts the probability of an incorrect base call. A higher Q score indicates a lower probability of error. For example, a Q score of 30 (Q30) corresponds to a 1 in 1,000 error rate, or 99.9% accuracy. The relationship between Q scores and accuracy follows a logarithmic scale, where each 10-point increase represents a tenfold decrease in error probability [10] [20].
The Path to Precision: Consensus Sequencing
Consensus sequencing is a strategy that sequences the same DNA molecule multiple times to generate a highly accurate consensus sequence. This approach effectively randomizes and cancels out stochastic errors inherent in single reads. Circular Consensus Sequencing (CCS), also known as HiFi sequencing from PacBio, implements this by circularizing DNA molecules and sequencing them multiple passes to produce highly accurate (99.8%) long reads [21] [20]. This method has revolutionized long-read genomics by providing both length and accuracy.
Technology Comparison: Accuracy and Error Profiles
Table 1: Sequencing Platform Performance Characteristics
| Platform/Technology | Read Length | Raw Read Accuracy | Consensus Accuracy (CCS) | Primary Error Type | Optimal Applications |
|---|---|---|---|---|---|
| PacBio HiFi (CCS) | 10-25 kb [1] [20] | ~90% (single pass) [20] | 99.9% (Q30) [1] [20] | Homopolymer indels [20] | Genome assembly, variant detection, haplotype phasing [20] |
| Oxford Nanopore (ONT) | Up to 4 Mb [1] | 95%-99% (R10.4 chemistry) [1] | >99% (with deep coverage) [10] | Systematic errors [10] | Direct RNA sequencing, structural variants, real-time analysis [1] |
| Illumina Short-Read | 50-300 bp [1] | 99.9% [1] | N/A | Substitution errors [20] | SNV detection, expression quantification, targeted sequencing [1] |
Table 2: Quantitative Performance Benchmarks from Recent Studies
| Performance Metric | PacBio HiFi | Oxford Nanopore | Illumina Short-Read |
|---|---|---|---|
| SNV Precision/Recall | >99.91% [20] | >99.9% (with Clair3/DeepVariant) [22] | >99.9% [20] |
| Indel Precision/Recall | 95.98% [20] | High (with deep learning callers) [22] | >99% [20] |
| Mapping Rate | Highest (97.5%) [20] | ~85% [23] | 94.8% [20] |
| Homopolymer Error Rate | 1 per 477 bp [20] | Improved with R10.4 chemistry [22] | Very low |
| Mismatch Rate | 1 per 13,048 bp [20] | Higher than short-read (context-dependent) [23] | 1 per 225,000 bp [20] |
Experimental Protocols for Assessing Accuracy
Circular Consensus Sequencing (CCS) Library Preparation
The following protocol for generating high-accuracy long reads has been optimized for PacBio systems [21] [20]:
DNA Fragmentation and Size Selection: High molecular weight (HMW) DNA is extracted and sheared to a tight size distribution around 15 kb using systems like the Megaruptor 3. This controlled fragmentation is crucial for optimizing polymerase read length and consensus accuracy.
Library Construction with Pre-extension: The sheared DNA is converted to a SMRTbell library via end-repair, A-tailing, and hairpin adapter ligation. A critical "pre-extension" step is employed where the polymerase extends without laser illumination. This eliminates polymerases on damaged templates before sequencing begins, significantly improving read length and yield.
Sequencing and Consensus Generation: The library is sequenced on PacBio Sequel IIe or Revio systems with collection times adjusted to maximize polymerase survival. The circularized molecules are sequenced multiple times (typically ≥10 passes), and CCS algorithms generate highly accurate consensus sequences from these subreads with calibrated quality scores.
Accuracy Validation and Benchmarking
To validate the accuracy of consensus sequences and quality scores, researchers employ these established methods [21] [20] [23]:
GIAB Benchmark Comparison: Sequence data is aligned to well-characterized human reference genomes from the Genome in a Bottle (GIAB) Consortium, such as HG002/NA24385. Precision and recall are calculated for single nucleotide variants (SNVs), insertions/deletions (indels), and structural variants against the validated benchmark variant set.
Umbilical Cord Blood Analysis: For somatic variant calling applications, sequencing data from umbilical cord blood (which has an exceedingly low number of true somatic variants due to its relatively young age) is analyzed. Bases that differ from the reference but are not at germline variant locations are counted as errors, providing a real-world measure of accuracy.
Read-to-Read Alignment: An independent method where reads are aligned to each other instead of a reference genome. This approach estimates error rates and identifies artifacts like molecular chimeras (0.5% in CCS reads) and low-quality base runs, providing orthogonal validation of sequence quality.
Visualizing Sequencing and Analysis Workflows
Circular Consensus Sequencing (CCS) Workflow
Diagram 1: CCS sequencing generates highly accurate long reads by sequencing circularized DNA molecules multiple times and deriving a consensus sequence from the subreads [21] [20].
TopoQual Error Correction and Quality Refinement
Diagram 2: The TopoQual algorithm uses partial order alignment and deep learning to polish consensus sequences and predict more accurate base quality scores [21].
The Scientist's Toolkit: Essential Research Reagents and Computational Tools
Table 3: Key Reagents and Computational Tools for Sequencing Accuracy Analysis
| Tool/Reagent | Type | Function | Application Context |
|---|---|---|---|
| TopoQual [21] | Software | Polishes CCS data using partial order alignments and deep learning | Corrects ~31.9% of errors in PacBio consensus sequences; validates base qualities up to q59 |
| MAS-ISO-seq/Kinnex [4] | Library Prep | Concatenates transcripts for efficient long-read RNA sequencing | Enables high-throughput scRNA-seq with isoform resolution; retains transcripts <500 bp |
| DeepVariant/Clair3 [22] | Variant Caller | Deep learning-based variant detection from sequencing data | Significantly outperforms traditional methods on ONT data; matches/exceeds Illumina accuracy |
| GIAB Reference Materials [20] [23] | Benchmark | Well-characterized human genome standards for validation | Provides ground truth for accuracy assessment across platforms and pipelines |
| SMRTbell Prep Kit [20] | Library Prep | Reagents for constructing circular sequencing libraries | Essential for PacBio HiFi sequencing with optimized adapter ligation |
| Nanoseq Pipeline [6] | Bioinformatics | Community-curated workflow for long-read RNA-seq data | Performs quality control, alignment, transcript discovery, and quantification |
The evolution of sequencing technologies, particularly through consensus methods like PacBio HiFi, has dramatically narrowed the accuracy gap between long-read and short-read platforms. While each technology maintains distinct error profiles—with long reads excelling in complex genomic regions and short reads providing exceptional base-level precision—the emergence of sophisticated computational tools like TopoQual and DeepVariant further enhances data quality [21] [22].
For researchers designing sequencing studies, the choice between platforms now depends less on raw accuracy alone and more on the specific genomic contexts of interest, required read lengths, and the complementarity of these technologies. The experimental protocols and benchmarking frameworks presented here provide a foundation for rigorous assessment of sequencing accuracy in diverse research applications, from basic transcriptome characterization to clinical diagnostics and drug development.
The field of genomic sequencing has undergone a revolutionary transformation with the advent of third-generation sequencing (TGS) technologies. Unlike their second-generation predecessors, which rely on amplified DNA fragments and produce short reads, TGS platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) enable single-molecule, real-time sequencing of long nucleic acid fragments. This evolution has fundamentally addressed one of the most significant initial limitations of TGS: high error rates. Through continuous technological refinement, TGS has progressed to offer remarkable fidelity while maintaining its inherent advantages for resolving complex genomic regions, characterizing structural variations, and providing full-length transcriptomic views. This guide objectively compares the performance of modern high-fidelity TGS with both short-read sequencing and earlier long-read approaches, providing researchers with critical insights for selecting appropriate sequencing strategies.
Historical Context and Technological Foundations
The Sequencing Technology Landscape
Next-generation sequencing (NGS) encompasses several technological generations that have progressively enhanced our ability to decode genetic information. First-generation sequencing, exemplified by Sanger's chain-termination method, provided accurate but low-throughput sequencing capabilities [24]. Second-generation sequencing (short-read technologies) from platforms like Illumina revolutionized genomics through massive parallel sequencing, offering high accuracy at reduced costs but producing fragments typically between 50-300 base pairs [24] [25]. These short reads struggle to resolve repetitive elements, structural variations, and complex genomic regions.
Third-generation sequencing emerged around 2011 with fundamentally different approaches [26]. PacBio's Single Molecule Real-Time (SMRT) technology and ONT's nanopore sequencing enabled the direct sequencing of single DNA or RNA molecules without amplification, producing reads that can span thousands to hundreds of thousands of bases [24] [26]. This technological leap came with an initial trade-off: early TGS platforms exhibited error rates substantially higher than Illumina's >99.9% base-calling accuracy [27] [25].
The High Error Rate Challenge
The initial limitations of TGS stemmed from their distinct sequencing chemistries. Early PacBio SMRT sequencing was prone to indels due to the instability of molecular machinery, while ONT's signal interpretation was complicated by adjacent base signal interference [27]. These technical challenges resulted in error rates that could reach 10-15% in some applications, posing significant obstacles for detecting single-nucleotide variants within the context of minimal genetic variation between individuals [24] [27].
The Path to High Fidelity: Technological Advancements
PacBio's HiFi Sequencing Breakthrough
Pacific Biosciences addressed accuracy challenges through the development of HiFi (High-Fidelity) sequencing. This approach uses circular consensus sequencing (CCS), where DNA molecules are sequenced repeatedly in a looped format. By generating multiple observations of each base, HiFi sequencing achieves accuracy exceeding 99.9% while maintaining read lengths of 10-25 kilobases [28] [24]. This technological advancement has made PacBio HiFi suitable for applications requiring both long reads and high accuracy, including variant detection, haplotype phasing, and assembly of complex genomes.
Nanopore's Accuracy Enhancements
Oxford Nanopore Technologies has progressively improved its sequencing accuracy through enhanced nanopore chemistries, motor enzymes, and base-calling algorithms. While early ONT platforms had error rates around 5-15%, recent developments have substantially improved performance [24] [6]. The SG-NEx project benchmarking demonstrated that ONT can now robustly identify major isoforms and detect complex transcriptional events, though it still trails PacBio in certain SNP calling applications [6].
Comparative Performance of Modern Sequencing Platforms
Table 1: Performance Comparison of Major Sequencing Technologies
| Platform | Read Length | Accuracy | Key Strengths | Primary Limitations |
|---|---|---|---|---|
| Illumina | 50-300 bp | >99.9% | High throughput, low cost per base, well-established bioinformatics | Short reads struggle with repeats and structural variants |
| PacBio HiFi | 10,000-25,000 bp | >99.9% | Long reads with high accuracy, excellent for structural variants and haplotype phasing | Higher cost per base, lower throughput than Illumina |
| PacBio Onso | 100-200 bp | High (SBB chemistry) | Targeted sequencing with binding chemistry | Higher cost compared to other targeted approaches |
| Oxford Nanopore | 10,000-30,000+ bp | Improved (recent platforms) | Ultra-long reads, direct RNA sequencing, portability | Higher error rates than HiFi, though improving |
Table 2: RNA Sequencing Protocol Comparisons (SG-NEx Benchmark)
| Protocol | Average Read Length | Throughput | 5'/3' Coverage | Best Applications |
|---|---|---|---|---|
| Illumina Short-Read | Fixed by protocol | Very high | Fragmentation biases | Gene-level expression, large sample numbers |
| PacBio Iso-Seq | Longest on average | High (with Kinnex) | Uniform coverage | Full-length isoform discovery, novel splicing |
| Nanopore Direct RNA | Long | Moderate | Higher at 3' end | Native RNA detection, modification analysis |
| Nanopore cDNA PCR | Long | Highest for Nanopore | Uniform coverage | Standard isoform expression profiling |
Experimental Evidence: Demonstrating Modern TGS Performance
Benchmarking Studies and Performance Metrics
Recent comprehensive benchmarks have quantitatively established the capabilities of modern TGS. The Singapore Nanopore Expression (SG-NEx) project, one of the most extensive comparisons of RNA sequencing protocols, found that long-read RNA sequencing more robustly identifies major isoforms compared to short-read approaches [6]. The study reported that PacBio IsoSeq generated the longest reads on average with uniform coverage across transcripts, while Nanopore cDNA sequencing achieved the highest throughput for long-read protocols [6].
Single-Cell RNA Sequencing Comparison
A systematic comparison of single-cell long-read and short-read sequencing demonstrated that both methods yield highly comparable results for standard gene expression analysis [4]. However, long-read sequencing provided the crucial advantage of isoform resolution, enabling the identification of 44,325 transcript isoforms in mouse retina cells, with 38% previously uncharacterized and 17% expressed exclusively in distinct cellular subclasses [29]. This study highlighted that while short-read sequencing provided higher sequencing depth, long-read sequencing allowed for identification of full-length transcripts and removal of technical artifacts [4].
Targeted Benchmarking of PacBio Kinnex
Recent evaluations of PacBio's high-throughput Kinnex kits revealed exceptionally strong concordance with Illumina data, with Pearson correlations exceeding 0.9 at the gene level and approaching 0.9 at the transcript level [16]. Importantly, the study found that "Illumina exhibited substantially higher inferential variability compared to Kinnex," with greater replicate-to-replicate fluctuations in transcript abundance estimates [16]. This demonstrates that modern TGS not only matches short-read accuracy but exceeds it in quantification consistency for complex isoforms.
Methodologies: Experimental Protocols for TGS Applications
PacBio HiFi Metagenomics Protocol
Metagenomics studies have particularly benefited from HiFi sequencing. The standard protocol involves:
- DNA Extraction: High-molecular-weight DNA extraction using kits optimized for long fragments
- Library Preparation: SMRTbell library construction with DNA repair, end-prep, and adapter ligation
- Size Selection: BluePippin or Circulomics size selection to enrich for longer fragments
- Sequencing: Loading on SMRT cells for circular consensus sequencing on Sequel IIe or Revio systems
- Data Processing: CCS read generation yielding HiFi reads with >99.9% accuracy [28]
This approach has demonstrated superior capability in recovering complete and coherent microbial genomes from complex microbiomes compared to both short-read and earlier long-read technologies [28].
Single-Cell Isoform Sequencing (Iso-Seq) Workflow
For comprehensive transcriptome profiling, the Iso-Seq protocol enables full-length transcript characterization:
- cDNA Synthesis: Full-length cDNA generation with template-switching reverse transcription
- PCR Optimization: Amplification with minimal bias using high-fidelity polymerases
- SMRTbell Library Preparation: Construction of libraries suitable for PacBio sequencing
- Size Selection: Fractionation to prioritize longer transcripts
- Sequencing: Single-molecule real-time sequencing capturing complete transcripts
- Bioinformatic Processing: CCS analysis, isoform clustering, and quantification [29] [16]
This methodology has been instrumental in revealing previously unannotated isoforms, with studies identifying approximately 40% novel transcripts not present in reference annotations [16].
Nanopore Direct RNA Sequencing Protocol
For native RNA analysis without cDNA conversion:
- RNA Quality Control: Assessment of RNA integrity number (RIN) >8.5
- Adapter Ligation: Poly(A) tail capture and adapter ligation
- Library Loading: Direct loading of RNA-library complexes onto flow cells
- Sequencing: Real-time sequencing through nanopores
- Base Calling: Signal processing to sequence while preserving modification information [6]
This approach uniquely enables direct detection of RNA modifications including N6-methyladenosine (m6A) alongside sequence information [6].
Visualization of Third-Generation Sequencing Workflows
PacBio SMRT Sequencing Technology
Third-Generation Sequencing Evolution
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents for Third-Generation Sequencing
| Reagent/Solution | Function | Application Examples |
|---|---|---|
| SMRTbell Libraries | Template for PacBio sequencing; enables circular consensus | HiFi sequencing, structural variant detection |
| MAS-ISO-seq/Kinnex Kits | Transcript concatenation for higher throughput | Single-cell isoform sequencing, full-length RNA-seq |
| Direct RNA Sequencing Kits | Native RNA sequencing without cDNA conversion | RNA modification analysis, epitranscriptomics |
| High-Molecular-Weight DNA Kits | Preservation of long DNA fragments | Metagenomics, genome assembly, structural variants |
| Barcoded Adapters | Sample multiplexing in single runs | Multi-sample experiments, cost reduction |
| Polymerase Binding Kits | Preparation of sequencing complexes | PacBio SMRT sequencing efficiency |
Third-generation sequencing has unequivocally evolved from its initial high-error state to become a high-fidelity technology that competes directly with short-read sequencing in accuracy while offering substantial advantages in resolving power. PacBio's HiFi sequencing now delivers >99.9% accuracy with read lengths of 10-25 kb, while Nanopore technologies continue to improve in both accuracy and read length capabilities. The choice between short-read and modern long-read sequencing now depends primarily on the specific research question rather than fundamental accuracy concerns. For applications requiring resolution of complex genomic regions, characterization of structural variants, detection of base modifications, or comprehensive transcript isoform analysis, third-generation sequencing offers unparalleled capabilities that continue to expand the frontiers of genomic research.
Strategic Application in Research: Choosing the Right Tool for Your Biological Question
For researchers and drug development professionals investigating gene expression profiles and single nucleotide polymorphisms (SNPs), short-read RNA sequencing has established itself as the cornerstone technology. Platforms like Illumina, Ion Torrent, and Element Biosciences generate sequences spanning tens to hundreds of base pairs, offering an unmatched combination of high accuracy, cost-effectiveness, and scalability [30]. While long-read technologies from PacBio and Oxford Nanopore Technologies excel at resolving complex isoform structures, the domain of high-throughput gene expression and SNP analysis remains powerfully addressed by short-read methodologies [6] [10]. This guide objectively compares the performance of short-read and long-read RNA sequencing, providing supporting experimental data to illustrate why short-read platforms continue to be the default choice for large-scale transcriptomic studies in drug discovery and basic research.
Technology Comparison: How Short-Reads and Long-Reads Measure Up
Core Technical Characteristics
The fundamental differences in technology architecture between short-read and long-read platforms create a natural division in their optimal applications.
Table 1: Fundamental Comparison of Short-Read and Long-Read RNA-Sequencing Technologies
| Feature | Short-Read cDNA-Seq | Long-Read cDNA-Seq |
|---|---|---|
| Representative Platforms | Illumina, Ion Torrent, Element Biosciences AVITI [10] | PacBio, Oxford Nanopore Technologies (ONT) [30] |
| Typical Read Length | Tens to hundreds of base pairs [30] | Thousands to hundreds of thousands of base pairs [10] |
| Key Strengths | Very high throughput, high accuracy (Q40+), cost-effective, scalable, well-understood bias and error profiles [30] [10] | Captures full-length transcripts, simplifies isoform discovery and fusion transcript detection [30] |
| Primary Limitations | Limited direct isoform detection, introduction of amplification biases [30] | Low to medium throughput, higher cost per sample, more complex data processing [30] |
Performance in Gene Expression and SNP Detection
Recent comparative studies quantify the performance gap in core applications. Short-read sequencing provides higher sequencing depth, which is critical for confidently detecting subtle gene expression changes and low-frequency SNPs [4]. In a 2025 study that sequenced the same 10x Genomics 3' cDNA using both Illumina and PacBio platforms, short-reads demonstrated a superior ability to recover more unique molecular identifiers (UMIs) per cell, a key metric for quantitative single-cell gene expression analysis [4].
Long-read sequencing, while transformative for isoform resolution, has not surpassed short-reads for pure gene-level quantification. The SG-NEx (Singapore Nanopore Expression) project, a comprehensive benchmark published in Nature Methods in 2025, found that while long-read protocols can robustly estimate gene expression, the massive throughput of short-read data makes it exceptionally reliable for this purpose [6]. For SNP detection, the high per-base accuracy of short-reads (often exceeding Q40 on modern platforms like the Element Biosciences AVITI System) is a decisive advantage for identifying single-nucleotide variants with high confidence [10].
Experimental Evidence: A Head-to-Head Comparison
Methodology of a Paired-Study
To ensure a fair comparison, researchers have designed experiments that sequence the same cDNA library with both short- and long-read technologies.
- Sample Preparation: A typical protocol begins with the conversion of RNA to cDNA, tagged with cell barcodes and UMIs. For example, one study used the 10x Genomics Chromium Single Cell 3' Reagent Kit (v3.1 Chemistry Dual Index) on patient-derived organoid cells [4].
- Library Splitting: The same pool of amplified, full-length cDNA is then split for two separate library preparations.
- Illumina (Short-Read) Library: The cDNA is enzymatically sheared to a target size of 200-300 bp. Following end repair, A-tailing, and adapter ligation, a sample index PCR is performed. Sequencing is done on an Illumina NovaSeq 6000 to achieve a high depth of ~300,000 reads per cell [4].
- PacBio (Long-Read) Library: The same cDNA is used for single-cell MAS-ISO-seq (Multiplexed Array Isoform Sequencing) library preparation. This involves a PCR step to remove template-switching oligo (TSO) artefacts, followed by directional assembly of cDNA segments into long concatenated arrays (10-15 kb) for efficient sequencing on a PacBio Sequel IIe system [4].
- Data Analysis: Reads are demultiplexed, aligned to the reference genome, and mapped to genes. For the comparison, molecules are matched by their cell barcode and UMI to enable a per-molecule cross-comparison [4].
Key Quantitative Findings from Direct Comparisons
This paired experimental design yields clear, data-driven results.
Table 2: Experimental Performance Data from a Paired Sequencing Study [4]
| Performance Metric | Illumina Short-Reads | PacBio Long-Reads | Implication for Researchers |
|---|---|---|---|
| Sequencing Depth | High (Target: ~300,000 reads/cell) | Lower (~2 million reads total per SMRT cell) | Short-reads offer greater depth for statistical power in DGE and SNP calling. |
| UMIs Recovered per Cell | Higher | Lower | Enables more precise quantification of transcript molecules in single-cell studies. |
| Transcript Length Bias | Recovered fewer transcripts <500 bp | Retained transcripts shorter than 500 bp | Long-reads can profile very short transcripts missed by standard short-read protocols. |
| Handling of Artefacts | Standard filtering | Stringent filtering of TSO-contaminated cDNA | Long-read library prep can remove specific artefacts, potentially purifying the data. |
| Gene Count Correlation | High correlation between methods | Correlation reduced after filtering long-read artefacts | Highlights that platform-specific processing impacts final gene expression matrices. |
The overarching finding is that both methods are highly comparable and recover a large proportion of cells and transcripts [4]. However, the higher throughput and UMI recovery of short-read sequencing make it particularly suited for studies where quantifying the expression levels of thousands of genes across many samples is the primary goal.
The Researcher's Toolkit for Short-Read RNA-Seq
Successful gene expression and SNP detection studies rely on a suite of trusted reagents and methodologies.
Table 3: Essential Research Reagent Solutions for Short-Read RNA-Seq
| Tool / Reagent | Function | Considerations for Experimental Design |
|---|---|---|
| Poly(A) Capture Beads | Enriches for polyadenylated mRNA by hybridization to oligo(dT) probes. | Not suitable for degraded RNA or non-polyA RNAs (e.g., some lncRNAs) [31]. |
| rRNA Depletion Kits | Reduces the ~80% of cellular RNA that is ribosomal, increasing informative reads. | More cost-effective for transcriptome coverage; assess off-target effects on genes of interest [32]. |
| Stranded Library Prep Kits | Preserves the original orientation of the transcript during cDNA synthesis. | Critical for identifying overlapping genes, novel RNAs, and accurate isoform assignment [32]. |
| Unique Molecular Identifiers (UMIs) | Short random sequences added to each molecule pre-amplification to correct for PCR bias. | Enables precise digital counting of transcripts, essential for single-cell RNA-seq [4]. |
| Size Selection Beads | Performs a solid-phase reversible immobilization (SPRI) to select for a specific cDNA fragment size. | Standard post-amplification clean-up and double-sided size selection are common in Illumina protocols [4]. |
Decision Workflows and Experimental Design
The choice between sequencing technologies is a fundamental step in experimental design. The following workflow diagram outlines the key decision points based on the primary research goal.
In the context of a broader comparison of RNA sequencing technologies, the evidence confirms that short-read sequencing remains the dominant force for high-throughput gene expression analysis and SNP detection. Its unparalleled throughput, high accuracy, and cost-efficiency make it the practical and powerful choice for transcriptomic studies in drug discovery, biomarker identification, and population-scale genomics [4] [30] [34]. While long-read sequencing opens up transformative possibilities for understanding transcriptome complexity, the quantitative strengths of short-reads ensure their continued central role in the molecular biologist's toolkit for years to come.
Long-read sequencing technologies have emerged as transformative tools for transcriptomics, enabling the direct observation of full-length RNA molecules. This capability is proving critical for discovering novel transcript isoforms and unraveling the complexity of gene regulation in health and disease. While short-read sequencing has been the workhorse for gene-level expression analysis, its limitations in resolving complete RNA structures have become increasingly apparent. This guide objectively compares the performance of long-read and short-read RNA sequencing technologies, supported by recent experimental data that highlight the unique advantages of long-read approaches for isoform-level analysis.
RNA sequencing has revolutionized how scientists study gene expression, providing an unbiased approach to gene detection and quantification [2]. For years, short-read sequencing has been the gold standard, offering high-throughput and cost-effective gene expression profiling [4]. However, a significant limitation persists: short reads (typically 100-200 base pairs) must be computationally assembled to approximate full transcripts, introducing ambiguity when resolving complex splicing patterns or distinguishing highly similar isoforms [35]. Long-read sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) directly address this limitation by sequencing full-length cDNA or RNA molecules in single reads, preserving exon connectivity and enabling direct observation of transcript structures [36] [2]. This capability is particularly valuable for understanding complex biological systems where alternative splicing generates multiple protein isoforms with distinct functions from a single gene.
Technical Comparison: Long-Read vs. Short-Read Sequencing
The fundamental differences between short-read and long-read technologies create distinct advantages and limitations for transcriptome analysis.
Table 1: Technical Comparison of RNA Sequencing Approaches
| Feature | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Read Length | 100-200 bp | 1,000 - 20,000+ bp |
| Isoform Resolution | Indirect inference through assembly | Direct observation of full-length isoforms |
| Primary Applications | Gene expression quantification, differential expression | Isoform discovery, alternative splicing analysis, fusion detection |
| Splice Junction Mapping | Ambiguous for complex genes | Precise determination of exon connectivity |
| Throughput | Very high | Moderate to high (increasing with newer platforms) |
| Error Profile | Low random errors (~0.1%) | Higher single-pass error rates, mitigated by circular consensus sequencing (HiFi) |
| Identification of Novel Features | Limited by read length | Comprehensive discovery of novel isoforms, exons, and gene fusions |
Key Advantages of Long-Read Sequencing
- Full-Length Transcript Coverage: Long reads can capture complete transcripts from 5' to 3' end in a single read, providing unambiguous isoform information [2] [35].
- Discovery of Novel Isoforms: Multiple studies have demonstrated long-read technologies identify tens of thousands of previously unannotated isoforms. Research on human whole blood identified approximately 90,000 novel isoforms using PacBio long-read RNA-seq [37].
- Resolution of Complex Loci: Genes with numerous alternative exons or long repetitive regions, which are challenging for short-read assembly, can be fully characterized with long reads [38].
- Phasing Capability: Long reads preserve haplotype information, enabling allele-specific expression analysis of isoforms [2].
Experimental Evidence: Performance Benchmarks
Recent large-scale benchmarking studies and targeted investigations have quantitatively compared the performance of long-read and short-read technologies for transcriptome analysis.
The LRGASP Consortium Benchmark
The Long-read RNA-Seq Genome Annotation Assessment Project (LRGASP) Consortium conducted a systematic evaluation of long-read RNA-seq methods for transcript identification and quantification [39]. This comprehensive effort generated over 427 million long-read sequences from human, mouse, and manatee samples using multiple protocols and sequencing platforms.
Table 2: LRGASP Performance Metrics for Transcript Detection
| Metric | cDNA-PacBio | cDNA-ONT | R2C2-ONT | CapTrap-PacBio |
|---|---|---|---|---|
| Read Length | Longest distributions | Moderate | Longest distributions | Moderate |
| Sequence Quality | High | Lower | High | High |
| Throughput (reads) | Moderate | ~10x higher than other methods | Moderate | Moderate |
| FSM Detection | High with Bambu, IsoQuant, FLAIR | Variable across tools | Not specified | Not specified |
| Novel Transcript Support | High full support for novel transcripts | Lower support for novel transcripts | Not specified | Not specified |
The consortium found that libraries with longer, more accurate sequences (such as cDNA-PacBio) produced more accurate transcripts than those with increased read depth, while greater read depth improved quantification accuracy [39]. For well-annotated genomes, tools based on reference sequences (including Bambu, FLAIR, FLAMES, and IsoQuant) demonstrated the best performance in detecting known transcripts with high percentages of full splice matches.
Direct Platform Comparison Studies
A focused study comparing single-cell long-read and short-read sequencing of the same 10x Genomics complementary DNA (cDNA) libraries found that both methods recovered a large proportion of cells and transcripts with highly comparable results [4]. However, platform-dependent cDNA library processing introduced specific biases:
- Short-read sequencing provided higher sequencing depth
- Long-read sequencing (PacBio MAS-ISO-seq) retained transcripts shorter than 500 bp and enabled removal of degraded cDNA contaminated by template switching oligos
- Filtering of artifacts identifiable only from full-length transcripts reduced gene count correlation between the two methods
The Singapore Nanopore Expression (SG-NEx) project provided additional insights through a systematic benchmark of Nanopore long-read RNA sequencing for transcript-level analysis in human cell lines [6]. This comprehensive resource compared five RNA-sequencing protocols across seven human cell lines and reported that:
- PCR-amplified cDNA sequencing generated the highest throughput among long-read protocols
- PacBio IsoSeq generated the longest reads on average
- Long-read protocols showed higher coverage at the 5' and 3' ends of transcripts compared to short-read RNA-seq
- Gene expression estimates from Nanopore long-read RNA-seq data showed low estimation error and high correlation with expected spike-in concentrations
Detailed Experimental Protocols
To illustrate the practical application of long-read sequencing for isoform discovery, we detail two key methodologies from recent studies.
Protocol 1: MAS-ISO-seq for Single-Cell Isoform Sequencing
The MAS-ISO-seq (Multiplexed Array isoform sequencing) method, now relabeled as Kinnex full-length RNA sequencing, was used to profile patient-derived clear cell renal cell carcinoma organoids [4].
Library Preparation Workflow:
- cDNA Synthesis: Full-length cDNA was generated using the 10x Genomics Chromium Single Cell 3' Reagent Kits (v3.1 Chemistry Dual Index).
- TSO Artefact Removal: Template-switching oligonucleotide (TSO) priming artefacts generated during cDNA synthesis were removed using PCR with a modified primer (MAS capture primer Fwd) to incorporate a biotin tag into desired cDNA products, followed by capture with streptavidin-coated MAS beads.
- Segment Assembly: Purified cDNA was processed with programmable segmentation adapter sequences in 16 parallel PCR reactions per sample, followed by directional assembly of amplified cDNA segments into linear arrays of 10-15 kb.
- Library Construction: MAS arrays were DNA damage repaired and nuclease treated to produce final single-cell MAS-ISO-seq libraries.
- Quality Control: Library quantity and quality were measured by Qubit 1X dsDNA High Sensitivity Kit and pulse-field capillary electrophoresis system Femto Pulse.
- Sequencing: Libraries were sequenced on PacBio Sequel IIe systems using 3.2 binding chemistry on 8M SMRT cells.
This protocol demonstrated the ability to retain transcripts shorter than 500 bp and remove a large proportion of truncated cDNA contaminated by TSO artefacts [4].
Protocol 2: Nanopore Amplicon Sequencing for Neuropsychiatric Risk Genes
A specialized approach for deeply profiling the RNA isoform repertoire of 31 high-confidence neuropsychiatric disorder risk genes in human brain utilized nanopore long-read amplicon sequencing [38].
Experimental Workflow:
- Sample Collection: Seven regions of post-mortem human brain were collected from five control individuals, encompassing transcriptionally divergent regions and those implicated in mental health disorders.
- Amplicon Design: Primers were designed to cover the full coding region of target genes, running from the first to the last exon where possible.
- Multiple Primer Strategy: For genes with alternative transcriptional initiation and termination exons, multiple primer sets were employed to profile as many potential alternative isoforms as possible.
- Sequencing: Amplified products were sequenced using Oxford Nanopore Technologies.
- Bioinformatic Analysis: The custom pipeline IsoLamp was developed specifically for isoform discovery from amplicon sequencing data, demonstrating superior performance in benchmarking studies compared to existing tools.
This approach identified 363 novel isoforms and 28 novel exons in neuropsychiatric risk genes, with genes such as ATG13 and GATAD2A showing most expression from previously undiscovered isoforms [38].
The Scientist's Toolkit: Essential Research Reagents
Successful long-read transcriptomics requires specialized reagents and computational tools. The following table details essential solutions for conducting long-read RNA sequencing studies.
Table 3: Essential Research Reagents and Tools for Long-Read Transcriptomics
| Category | Specific Products/Tools | Function/Application |
|---|---|---|
| Library Prep Kits | PacBio Iso-Seq Express 2.0, ONT PCR-cDNA Kit | Convert RNA to sequencing-ready libraries with optimized protocols for full-length transcript capture |
| Spike-In Controls | SIRV Sets, ERCC RNA Spike-In Mixes | Assess technical performance, quantify detection limits, and normalize across experiments |
| Quality Control | Agilent 4200 TapeStation, Qubit dsDNA HS Assay | Evaluate RNA integrity, cDNA quality, and final library quantification before sequencing |
| Sequencing Platforms | PacBio Revio/Sequel IIe, ONT PromethION/P2 Solo | Generate long-read data with platform-specific advantages in read length and accuracy |
| Bioinformatics Pipelines | IsoLamp, Bambu, FLAIR, IsoQuant, TALON | Process raw data, discover novel isoforms, and quantify transcript expression |
| Reference Annotations | GENCODE, RefSeq, CHM13 T2T | Provide baseline transcript models for comparison and novel isoform classification |
| Validation Tools | SQANTI3, Isoseq v4.0, Pigeon | Perform quality control of long-read defined transcriptomes and classify full-length isoforms |
Application Highlights: Transforming Research Insights
Long-read sequencing has enabled groundbreaking discoveries across multiple biological domains by revealing previously inaccessible transcriptomic complexity.
Cancer Research
Application of PacBio long-read sequencing to breast cancer samples identified thousands of previously unannotated transcripts, with approximately 30% affecting protein-coding exons and predicted to alter protein localization and function [40]. The study further identified 3,059 breast tumor-specific splicing events, 35 of which were significantly associated with patient survival. Notably, 21 of these survival-associated events were absent from GENCODE annotations, demonstrating that clinically relevant splicing events remain undiscovered without long-read technologies.
Neuroscience
Comprehensive profiling of 31 neuropsychiatric risk genes in human brain revealed unprecedented isoform diversity, with the greatest complexity detected in the schizophrenia risk gene ITIH4 [38]. Mass spectrometry confirmation of a novel exon skipping event in ITIH4 suggested a new regulatory mechanism for this gene in the brain. For genes including ATG13 and GATAD2A, most expression was from previously undiscovered isoforms, fundamentally changing our understanding of these genes' expression in the brain.
Clinical Diagnostics
Research on chronic lymphocytic leukemia (CLL) samples using long-read single-cell RNA-seq with MAS-seq informed subclonal evolution patterns that may guide patient-specific therapies [41]. The ability to resolve full-length transcript isoforms at single-cell resolution provides unprecedented insight into tumor heterogeneity and cellular states in cancer progression.
Long-read sequencing technologies have fundamentally transformed transcriptomics by enabling direct observation of full-length RNA molecules. The evidence from recent benchmarks and application studies consistently demonstrates that long-read approaches provide unparalleled capabilities for isoform discovery, characterization of alternative splicing, and detection of novel transcripts. While short-read sequencing remains valuable for high-throughput gene expression quantification, long-read technologies excel in applications requiring complete transcript structure resolution.
As sequencing costs decrease and analytical methods mature, long-read RNA sequencing is positioned to become the new standard for comprehensive transcriptome analysis. For researchers and drug development professionals, embracing these technologies now provides a competitive advantage in understanding the complex transcriptional regulation underlying development, cellular function, and disease mechanisms.
Single-cell RNA sequencing (scRNA-seq) has emerged as a revolutionary technology in cancer research, providing unprecedented resolution to dissect the complex cellular architecture of tumors. Unlike traditional bulk RNA sequencing, which averages gene expression across thousands to millions of cells, scRNA-seq enables researchers to profile transcriptomes at individual cell resolution [42] [43]. This technological advancement is particularly crucial for understanding tumor heterogeneity—a fundamental characteristic of cancer that drives progression, metastasis, and therapy resistance [44] [45]. By revealing distinct cell subpopulations, rare cell types, and continuous transitional states within tumors, scRNA-seq provides unique insights into the molecular mechanisms governing cancer biology that were previously obscured in bulk analyses [46] [47].
The application of scRNA-seq in oncology aligns with the broader thesis comparing short-read and long-read sequencing technologies. While long-read sequencing excels in detecting isoform diversity and structural variants, high-throughput short-read scRNA-seq platforms have become the dominant approach for characterizing cellular heterogeneity due to their superior cell throughput, cost-effectiveness, and robust quantitative capabilities for gene expression quantification [42] [48]. This guide will objectively compare the performance of leading scRNA-seq technologies, their experimental frameworks, and their applications in resolving tumor heterogeneity and cell states, providing researchers with practical insights for selecting appropriate methodologies for their cancer studies.
Key scRNA-seq Platforms and Methodologies
scRNA-seq technologies have evolved significantly since their inception in 2009, with current methods primarily classified into two categories: full-length transcript sequencing approaches and 3′/5′-end transcript counting methods (tag-based) [46] [49]. Full-length methods such as SMART-seq2 provide uniform transcript coverage, enabling detection of alternative splicing, sequence variants, and allele-specific expression [46] [50]. In contrast, tag-based methods like those employed in 10x Genomics, Drop-seq, and inDrop focus on 3′ or 5′ transcript ends combined with unique molecular identifiers (UMIs) to minimize amplification bias, providing more quantitative gene expression data with higher throughput and lower cost [46] [50] [49].
From an implementation perspective, scRNA-seq platforms utilize either plate-based or droplet-based microfluidics for single-cell isolation [48]. Plate-based methods (e.g., Fluidigm C1, SMART-seq2) typically process fewer cells (96-800 cells per run) but offer higher sequencing depth and better detection of lowly-expressed genes [50]. Droplet-based systems (e.g., 10x Genomics Chromium, Drop-seq, inDrop) can profile thousands to tens of thousands of cells in a single experiment, making them ideal for comprehensive characterization of heterogeneous tissues like tumors [42] [46]. The choice between these approaches involves trade-offs between cell throughput, gene detection sensitivity, transcript coverage, and cost that must be carefully considered based on research objectives.
Comparative Performance of Major scRNA-seq Technologies
Table 1: Comparison of Major scRNA-seq Technologies and Platforms
| Technology | Read Coverage | Throughput (Cells) | Amplification Method | UMI Usage | Key Applications in Cancer Research |
|---|---|---|---|---|---|
| SMART-seq2 | Full-length | 102-103 | PCR-based (template switching) | No | Alternative splicing analysis, mutation detection in individual cells [46] [50] |
| 10x Genomics Chromium | 3' counting | 103-105 | PCR with template switching | Yes | Large-scale tumor heterogeneity studies, immune cell profiling [42] [46] |
| Drop-seq | 3' counting | 103-105 | PCR amplification | Yes | Cost-effective population screening, initial tumor characterization [46] [49] |
| CEL-seq2 | 3' counting | 103-104 | In vitro transcription (IVT) | Yes | High quantification accuracy, sensitive for low-abundance transcripts [46] [50] |
| MARS-seq | 3' counting | 103-104 | In vitro transcription (IVT) | Yes | Automated processing, immune cell heterogeneity [46] |
Table 2: Quantitative Performance Metrics Across Platforms
| Technology | Cells per Run | Cost per Cell | Gene Detection Sensitivity | Doublet Rate | Technical Noise |
|---|---|---|---|---|---|
| 10x Genomics | 10,000 (standard) | ~$0.50 | High (compared to other droplet methods) | Low with proper loading | Low [46] |
| Drop-seq | 10,000+ | ~$0.10 | Moderate | Moderate | Moderate [46] |
| inDrop | 10,000+ | ~$0.25 | Moderate | Moderate | Moderate [46] |
| MARS-seq2.0 | 8,000-10,000 | ~$0.10 | High with low background (2%) | Very low (<0.2%) | Low [46] |
| SMART-seq2 | 96-384 (plate-based) | Higher than droplet methods | Highest (full-length transcripts) | Low | Low with proper amplification [46] [50] |
The performance comparisons reveal that droplet-based methods generally provide the best balance of throughput and cost for large-scale tumor heterogeneity studies, with 10x Genomics offering superior sensitivity and lower technical noise [46]. Meanwhile, full-length methods like SMART-seq2 remain valuable for focused studies requiring comprehensive transcript information from smaller cell numbers [50]. Recent advancements such as MARS-seq2.0 have achieved remarkable reductions in both cost (sixfold reduction to $0.10 per cell) and background noise (2%), making high-quality scRNA-seq more accessible [46].
Experimental Framework: From Sample Processing to Data Analysis
Standardized Workflow for Tumor scRNA-seq
The typical scRNA-seq workflow begins with sample acquisition and processing, where tumor tissues are dissociated into single-cell suspensions [44] [50]. This critical step requires careful optimization as dissociation methods and temperature can induce artificial stress responses that alter transcriptional profiles [49]. For instance, dissociation at 4°C instead of 37°C minimizes heat shock protein induction, preserving more authentic expression patterns [49]. Single-cell isolation then follows using methods such as fluorescence-activated cell sorting (FACS), microfluidic chips, or droplet-based encapsulation [50] [48]. For tumor tissues with difficult dissociation properties, single-nucleus RNA sequencing (snRNA-seq) provides an alternative approach that minimizes dissociation artifacts and enables work with frozen specimens [49].
Following cell isolation, the library preparation phase involves cell lysis, reverse transcription with barcoded primers, cDNA amplification, and library construction [44] [46]. Reverse transcription typically employs oligo(dT) primers to capture polyadenylated RNA, with template-switching mechanisms (e.g., SMART technology) often used to generate full-length cDNA [50] [49]. Amplification is achieved either through PCR-based methods (e.g., SMART-seq2) or in vitro transcription (e.g., CEL-seq2), with the latter providing linear amplification that reduces technical noise [46] [50]. The incorporation of unique molecular identifiers (UMIs)—short random barcodes that label individual mRNA molecules—enables accurate transcript quantification by correcting for PCR amplification bias [44] [50]. The final sequencing step predominantly utilizes Illumina short-read platforms, providing the high throughput needed for profiling thousands of cells simultaneously [50].
Bioinformatic Analysis Pipeline
Table 3: Essential Computational Tools for scRNA-seq Data Analysis
| Analysis Step | Tool Options | Primary Function | Considerations for Tumor Samples |
|---|---|---|---|
| Quality Control | FastQC, Cell Ranger | Assess sequence quality, remove low-quality cells | Tumor cells may have higher mitochondrial content; adjust thresholds accordingly |
| Normalization | Seurat, Scanpy | Remove technical biases (sequencing depth, amplification efficiency) | Address elevated zeros in tumor data (dropout events) |
| Feature Selection | Seurat, Scanpy | Identify highly variable genes | Prioritize genes beyond standard housekeeping genes |
| Dimension Reduction | PCA, t-SNE, UMAP | Visualize high-dimensional data in 2D/3D | Can reveal malignant cell clusters and transitional states |
| Clustering | Seurat, Scanpy | Identify distinct cell populations | Over-clustering may help identify rare tumor subpopulations |
| Trajectory Inference | Monocle, PAGA, Slingshot | Reconstruct cellular developmental paths | Map tumor evolution and EMT transitions |
| Differential Expression | Seurat, MAST | Identify marker genes between conditions | Distinguish driver from passenger mutations in subclones |
The computational analysis of scRNA-seq data presents unique challenges due to technical artifacts, high dimensionality, and sparsity inherent to single-cell measurements [50] [48]. The standard pipeline begins with quality control to remove damaged cells, empty droplets, and multiplets (droplets containing more than one cell) [48]. Normalization follows to address technical variations in cDNA capture and amplification efficiency, with methods specifically designed to handle the excess zeros characteristic of single-cell data [50] [48]. Feature selection identifies highly variable genes that drive heterogeneity, reducing dimensionality for subsequent analyses [48].
Dimensionality reduction techniques like PCA, t-SNE, and UMAP enable visualization of cell relationships in two or three dimensions, revealing population structures [48]. Clustering algorithms then partition cells into distinct groups representing putative cell types or states, with resolution parameters significantly impacting the ability to identify rare subpopulations relevant in cancer [45] [48]. For studying dynamic processes like tumor progression or drug resistance, trajectory inference methods (e.g., Monocle, PAGA) reconstruct cellular paths through transcriptomic space, pseudo-temporal ordering of cells along differentiation or transition trajectories [48]. Additional specialized analyses include cell-cell communication inference through ligand-receptor interactions and copy number variation (CNV) estimation to distinguish malignant from non-malignant cells [45] [48].
Research Applications: Dissecting Tumor Heterogeneity and Microenvironment
Resolving Cancer Cell States and Plasticity
scRNA-seq has fundamentally advanced our understanding of intra-tumoral heterogeneity by revealing distinct cancer cell states within individual tumors [45]. Rather than discrete subtypes, malignant cells often exist along continuous phenotypic spectra, co-opting developmental programs like epithelial-mesenchymal transition (EMT) and differentiation hierarchies [45]. In glioblastoma, for instance, scRNA-seq has uncovered simultaneous coexistence of multiple cellular states—including stem-like, progenitor-like, and differentiated states—with functional implications for treatment resistance [45]. Similarly, in melanoma, cells exist along an axis from dormant, slow-cycling AXL-high states to proliferative MITF-high states, with the former conferring resistance to MAPK inhibitors [45].
The plasticity between these cell states represents a key mechanism of tumor adaptation and therapeutic evasion [45] [47]. Breast cancer tumors have been shown to contain cells corresponding to all molecular subtypes (basal, luminal A, luminal B, HER2) simultaneously, explaining how tumors can adapt to selective pressures [45]. This plasticity often follows principles of normal tissue homeostasis, with slow-cycling stem-like cells giving rise to rapidly proliferating progenitors that subsequently differentiate [45]. The ability to profile these dynamic transitions at single-cell resolution provides unprecedented opportunities to identify key regulators of cell state plasticity that could be targeted to limit tumor evolution and overcome treatment resistance.
Characterizing the Tumor Microenvironment
Beyond malignant cells, scRNA-seq has revolutionized our understanding of the tumor microenvironment (TME), revealing extraordinary complexity in immune and stromal compartments [44] [48]. The comprehensive immune cell profiling enabled by scRNA-seq has identified previously unappreciated immune subpopulations with clinical relevance—for instance, a small subset of CD8+ T cells associated with favorable response to adaptive cell transfer immunotherapy in melanoma, and regulatory T lymphocytes linked to poor prognosis in liver cancer [42]. In non-small cell lung cancer, a high proportion of active CD8+ T lymphocytes correlates with better outcomes, while specific macrophage subpopulations promote immunosuppression [42] [48].
The application of scRNA-seq in immuno-oncology has been particularly transformative, enabling detailed characterization of mechanisms underlying response and resistance to immune checkpoint blockade (ICB) therapy [48]. Studies comparing pre- and post-treatment samples have identified T cell populations predictive of ICB response, such as TCF7+CD8+ T cells that correlate with positive outcomes to anti-PD-1 treatment [48]. Similarly, the dynamic changes in myeloid cell composition and macrophage polarization states during therapy provide insights into alternative resistance mechanisms [48]. These findings not only advance our fundamental understanding of tumor immunology but also open avenues for developing novel immunotherapeutic strategies and biomarkers for treatment selection.
Essential Research Tools and Reagents
Table 4: Essential Research Reagent Solutions for scRNA-seq Experiments
| Reagent/Category | Specific Examples | Function in scRNA-seq Workflow | Technical Considerations |
|---|---|---|---|
| Cell Viability Kits | Propidium iodide, DAPI, Calcein AM | Distinguish live/dead cells during sorting | Critical for data quality; dead cells increase background noise |
| Cell Sorting Matrix | FACS buffers, BSA, EDTA | Maintain cell viability during isolation | Viscosity and composition affect sorting efficiency and cell integrity |
| Dissociation Enzymes | Collagenase, trypsin, liberase | Tissue dissociation to single cells | Enzyme choice and incubation time impact cell surface epitopes and RNA quality |
| Single-Cell Barcoding | 10x Barcoded Gel Beads, Drop-seq Beads | Cell and transcript indexing | Barcode complexity must exceed cell numbers to ensure uniqueness |
| Reverse Transcriptase | SmartScribe, Maxima H- | cDNA synthesis from single-cell RNA | High processivity and low RNase H activity improve yield |
| cDNA Amplification | KAPA HiFi HotStart, Advantage 2 | Whole transcriptome amplification | Faithful amplification minimizes technical bias in representation |
| Library Prep Kits | Nextera XT, Illumina Tagmentation | Sequencing library preparation | Size selection critical for removing primer dimers and artifacts |
| UMI Reagents | Custom UMI primers, commercial UMI sets | Unique molecular identifiers | UMI length (6-10 bp) must provide sufficient diversity for transcript population |
The successful implementation of scRNA-seq experiments depends critically on appropriate selection of research reagents and tools. Cell viability maintenance throughout the isolation process is paramount, as RNA degradation in dead cells significantly compromises data quality [50] [49]. The choice of tissue dissociation enzymes must balance efficiency with preservation of cell surface markers and transcriptional integrity [49]. For barcoding, commercial solutions like 10x Genomics' barcoded gel beads provide integrated solutions that ensure sufficient barcode diversity (3.6 million unique barcodes) to confidently label individual cells in large experiments [42] [46].
Enzyme selection for reverse transcription and amplification substantially impacts library quality and coverage bias. Reverse transcriptases with high processivity and template-switching activity (e.g., Maxima H-) improve cDNA yield, especially for long transcripts [50] [49]. PCR enzymes with high fidelity (e.g., KAPA HiFi) minimize amplification errors during library construction [50]. The incorporation of UMIs is now considered essential for accurate transcript quantification, with 6-8 bp random sequences providing sufficient diversity (46-48 = 4,096-65,536 possible sequences) to uniquely tag the approximately 100,000-1,000,000 mRNA molecules typically present in a single cell [50] [49].
scRNA-seq represents a powerful application of short-read sequencing technology that addresses fundamental questions in cancer biology inaccessible to bulk sequencing approaches. While long-read sequencing technologies provide advantages for characterizing isoform diversity and structural variants, the high throughput, quantitative accuracy, and cost-effectiveness of short-read scRNA-seq platforms have established them as the preferred method for large-scale single-cell transcriptomic studies [42] [48]. The ability to profile thousands of individual cells within heterogeneous tumors has revealed unprecedented insights into cancer cell states, tumor evolution, and microenvironmental interactions that underlie disease progression and treatment resistance.
As scRNA-seq technologies continue to evolve, emerging trends include multi-omic integrations (combining transcriptome with epigenome, proteome, or genome information from the same single cells), spatial transcriptomics (preserving spatial context in tissue sections), and computational methods for reconstructing lineage relationships and cellular dynamics [47] [48]. These advances, coupled with decreasing costs and increasing automation, promise to further transform cancer research and clinical practice. The strategic selection of appropriate scRNA-seq platforms and methodologies—whether droplet-based for large-scale heterogeneity studies or plate-based for focused investigations of transcriptional mechanisms—will remain essential for maximizing biological insights while efficiently utilizing resources. Through continued technological refinement and innovative application, scRNA-seq will undoubtedly play an increasingly central role in advancing our understanding of tumor biology and developing more effective cancer therapeutics.
The modern drug discovery pipeline is a high-stakes endeavor, taking approximately 10-15 years and costing between $900 million to over $2 billion per drug, with startling attrition rates in clinical trials due to unforeseen pharmacokinetics and toxicity issues [51]. In this challenging landscape, RNA sequencing technologies have emerged as transformative tools, enabling researchers to dissect cellular mechanisms at unprecedented resolution. The fundamental choice between short-read sequencing (e.g., Illumina) and long-read sequencing (e.g., PacBio, Oxford Nanopore) represents a critical strategic decision that influences every stage of drug development, from initial target identification to understanding drug resistance mechanisms.
While short-read RNA sequencing has dominated transcriptomic studies for years, providing high-throughput and high-quality gene-level information [4], it faces inherent limitations in capturing full-length transcript information and resolving complex splicing events [52]. Long-read RNA sequencing technologies overcome these limitations by enabling end-to-end sequencing of full-length transcripts, opening new avenues for investigating RNA species and features that cannot be reliably interrogated by short-read methods [8]. This technological evolution is particularly relevant for drug discovery, where understanding transcriptomic alterations that drive tumorigenesis, including splicing events, alternative polyadenylation, and open reading frames, can provide crucial insights for therapeutic development [52].
Technology Comparison: Short-Read vs. Long-Read Sequencing
Fundamental Technical Differences
The core distinction between short-read and long-read RNA sequencing methodologies lies in their approach to transcript capture and sequencing. Short-read sequencing relies on sequencing short fragments of cDNA, which are then aligned to a reference genome [52]. These methods typically provide partial transcript coverage either from the 3' or 5' end, with reads fixed to an exact length [4]. In contrast, long-read sequencing technologies span entire transcripts, allowing for direct detection of full-length isoforms and more accurate identification of splicing events [52].
For short-read platforms like Illumina NovaSeq 6000, library preparation involves enzymatic shearing of cDNA to target sizes of 200-300bp, followed by end repair, adapter ligation, and sample index PCR [4]. This approach provides high sequencing depth but sacrifices information about complete transcript structures. Long-read platforms like PacBio Sequel IIe utilize methods such as MAS-ISO-seq (now relabeled as Kinnex full-length RNA sequencing), which involves concatenating full-length transcripts into longer fragments that can be sequenced and later bioinformatically decomposed to original transcripts [4]. Oxford Nanopore Technologies' PromethION platform sequences native RNA or cDNA directly, avoiding amplification steps and preserving modification information [6].
Performance Comparison in Drug Discovery Applications
Table 1: Comparative Performance of Short-Read and Long-Read RNA Sequencing Technologies
| Parameter | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Transcript Coverage | Partial (3' or 5' end); read length fixed | Full-length; captures complete transcript structures |
| Isoform Resolution | Limited; inferential based on splice junctions | Direct detection of alternative isoforms |
| Sequencing Accuracy | High base-level accuracy | Historically lower, but significantly improved with recent advancements [52] |
| Throughput | Very high | Increasingly competitive, especially with PacBio Kinnex and ONT PromethION |
| Gene Expression Quantification | Robust for gene-level expression [4] | Comparable correlation (Pearson >0.9) at gene level; approaching 0.9 at transcript level [16] |
| Novel Transcript Discovery | Limited by read length | Superior; identifies ~40-50% novel transcripts not in reference databases [16] [37] |
| Single-Cell Applications | Well-established with 10x Genomics | Compatible with same single-cell barcoding systems [4] |
| Detection of Complex Events | Challenging for fusion genes, complex splicing | Excellent for fusion transcripts, allele-specific splicing, RNA modifications [6] |
Table 2: Performance Metrics from Recent Comparative Studies
| Metric | Short-Read (Illumina) | PacBio Long-Read | Nanopore Long-Read |
|---|---|---|---|
| Gene-Level Correlation | Reference | >0.9 Pearson correlation [16] | High correlation with spike-ins [6] |
| Transcript-Level Correlation | Limited by multi-mapping reads | ~0.9 Pearson correlation [16] | Protocol-dependent |
| Inferential Variability | Substantially higher replicate-to-replicate fluctuations [16] | Consistent quantification across replicates [16] | Intermediate |
| Full-Splice-Match Reads | Lower due to fragmentation | Highest in PCR-amplified cDNA and PacBio IsoSeq [6] | Varies by protocol |
| Coverage Uniformity | Biased toward 3' end in 3' mRNA-Seq | Uniform across transcript in PCR-amplified cDNA [6] | 3'-biased in direct RNA [6] |
Recent rigorous benchmarking studies demonstrate that both methods render highly comparable results and recover a large proportion of cells and transcripts [4]. However, platform-dependent cDNA library processing and data analysis steps introduce distinct biases. A 2025 study featuring one of the largest PacBio long-read RNA-seq datasets sample-matched with Illumina short-read RNA-seq found that "PacBio and Illumina quantifications were strongly concordant" with "Pearson correlations exceeding 0.9 at the gene level and approaching 0.9 at the transcript level" [16]. Notably, the study observed that "Illumina exhibited substantially higher inferential variability compared to Kinnex, with greater replicate-to-replicate fluctuations of estimated transcript abundances from the short-reads, whereas Kinnex demonstrated consistent quantification for the same transcripts" [16].
Experimental Protocols and Methodologies
Library Preparation Workflows
Short-read library preparation typically begins with RNA extraction, followed by either poly(A) selection for mRNA enrichment or ribosomal RNA depletion for whole transcriptome analysis. For 3' mRNA-Seq protocols like QuantSeq, library preparation initiates with oligo(dT) priming, generating one fragment per transcript, which streamlines the process and enables gene expression profiling even at low sequencing depths of 1-5 million reads per sample [53]. For whole transcriptome short-read sequencing, cDNA synthesis uses random primers, distributing reads across the entire transcript but requiring higher read depth to provide sufficient coverage [53].
Long-read library preparation varies by platform. For PacBio Iso-Seq, the process typically involves converting RNA to cDNA, then ligating SMRTbell adapters for sequencing [37]. The MAS-ISO-seq protocol (commercialized as Kinnex) includes a step to remove template switching oligonucleotide (TSO) artifacts generated during cDNA synthesis, followed by incorporation of programmable segmentation adapter sequences and directional assembly of amplified cDNA segments into linear arrays [4]. For Oxford Nanopore Technologies, three main protocols exist: PCR-amplified cDNA sequencing (highest throughput, least input RNA), amplification-free direct cDNA sequencing, and direct RNA sequencing (captures RNA modifications) [6].
Single-Cell RNA Sequencing Protocols
Single-cell RNA sequencing (scRNA-seq) has become particularly valuable in drug discovery for dissecting cellular heterogeneity in complex tissues and tumors. Both short-read and long-read technologies can be applied to the same single-cell barcoded libraries. A typical workflow involves using the 10x Genomics Chromium platform to partition cells into nanoliter-scale gel beads-in-emulsion (GEMs), where reverse transcription occurs with barcoded oligo-dT primers [4]. All cDNAs within a GEM share a common barcode, enabling single-cell resolution. The same single-cell full-length cDNA generated using 10x Genomics Chromium can be used to prepare both Illumina and PacBio sequencing libraries, allowing direct comparison of the technologies [4].
Workflow Comparison: Short-Read vs. Long-Read RNA Sequencing
Applications Across the Drug Discovery Pipeline
Target Identification and Validation
In target identification, scRNA-seq is crucial for identifying genes linked to specific cell types or novel states involved in disease, aiding in the discovery of potential drug targets [51]. The technology's ability to resolve cellular heterogeneity enables researchers to pinpoint disease-relevant cell populations and identify potential therapeutic targets specific to those populations. A 2024 retrospective analysis of known drug target genes demonstrated that targets with cell type-specific expression in disease-relevant tissues are more likely to progress successfully from Phase I to Phase II clinical trials [51].
Long-read sequencing enhances this process by providing isoform-resolution data, revealing previously unannotated transcripts in disease-relevant genes. In a study focusing on 556 unique genes causally implicated in early onset and syndromic epilepsy, Iso-Seq detected 4,341 unique novel transcripts, predicting 1,978 unique open reading frames (ORFs) [16]. Mass spectrometry subsequently identified 514 peptides unique to these predicted amino acid sequences, providing evidence for the translation of 745 transcripts (17.2%) with 356 unique ORFs (18.0%) [16]. This finding is particularly significant as "many of these genes belong to the druggable genome; KCNQ2 & STXBP1, are currently included in gene therapy trials listed on ClinicalTrials.gov" [16].
Lead Optimization and Mechanism of Action Studies
During lead optimization, understanding a compound's mechanism of action is critical. Traditional drug screening relies on general readouts like cell viability or marker expression, lacking comprehensive detail [51]. scRNA-seq enables detailed cell-type-specific gene expression profiles, essential for understanding drug mechanisms [51]. High-throughput screening now incorporates scRNA-seq for multi-dose, multiple experimental conditions, and perturbation analyses, providing richer data that support comprehensive insights into cellular responses, pathway dynamics, and potential therapeutic targets [51].
Long-read technologies provide additional depth by characterizing isoform-specific responses to therapeutic compounds. The ability to detect alternative splicing changes and isoform switching in response to treatment can reveal subtle mechanisms of drug action that would be missed by gene-level expression analysis. Furthermore, the identification of allele-specific splicing events—averaging 88 significant events per sample in one study of 202 Human Pangenome Reference Consortium (HPRC) Kinnex datasets, with 46% involving unannotated junctions—provides additional resolution for understanding genetic determinants of drug response [16].
Biomarker Discovery and Patient Stratification
Biomarker identification has been transformed by single-cell and long-read technologies. Traditionally, biomarkers were identified using various techniques, with bulk transcriptomics historically used to identify cancer biomarkers [51]. However, bulk approaches fail to capture cell population complexity. scRNA-seq has advanced this field by defining more accurate biomarkers, such as those in colorectal cancer, leading to new classifications with subtypes distinguished by unique signaling pathways, mutation profiles, and transcriptional programs [51].
Long-read sequencing enhances biomarker discovery by revealing isoform-level biomarkers that may be more specific than gene-level markers. In whole blood studies—particularly relevant for minimally invasive diagnostic applications—long-read RNA sequencing identified approximately 90,000 novel isoforms using the GRCh38 reference, representing 47% of total isoforms detected [37]. This comprehensive isoform catalog in accessible tissues like blood enables the development of more precise diagnostic and prognostic biomarkers based on specific transcript variants rather than overall gene expression.
Understanding Drug Resistance Mechanisms
Drug resistance remains a major challenge in oncology and other therapeutic areas. Long-read RNA sequencing provides unique advantages for deciphering resistance mechanisms through its ability to detect fusion transcripts, alternative isoforms associated with resistance, and RNA modifications that may influence drug sensitivity. The Singapore Nanopore Expression (SG-NEx) project demonstrated long-read RNA sequencing's capability to profile full-length fusion transcripts, alternative isoforms, and N6-methyladenosine (m6A) RNA modifications from direct RNA sequencing data [6].
In cancer research, long-read RNA-seq has emerged as a powerful technique for understanding the transcriptomic alterations that drive tumorigenesis [52]. It enables detection of splicing events, alternative poly(A) adenylation, and open reading frames that are often identified inefficiently or missed by short-read RNA-seq [52]. Furthermore, it offers insights into transcriptome-wide changes that may have implications for drug resistance, tumor progression, and metastasis [52].
Essential Research Reagents and Platforms
Table 3: Research Reagent Solutions for RNA Sequencing in Drug Discovery
| Reagent/Platform | Function | Application in Drug Discovery |
|---|---|---|
| 10x Genomics Chromium | Partitions cells into GEMs for single-cell barcoding | Enables single-cell resolution in both short-read and long-read workflows [4] |
| PacBio Iso-Seq Express 2.0 | cDNA synthesis for long-read sequencing | Full-length transcript identification for target validation [37] |
| Parse Biosciences Evercode v3 | Combinatorial barcoding for scRNA-seq | Large-scale perturbation studies (e.g., 10M cells, 1,092 samples) [51] |
| Oxford Nanopore cDNA-PCR Kit | PCR-amplified cDNA library preparation | High-throughput long-read sequencing with minimal input [6] |
| Lexogen QuantSeq | 3' mRNA-Seq library preparation | Cost-effective gene expression screening for large compound libraries [53] |
| NEBNext Poly(A) mRNA Magnetic Isolation | mRNA enrichment from total RNA | Target preparation for both short-read and long-read sequencing [52] |
| MAS-ISO-seq for 10x Genomics | Concatemerization for enhanced throughput | High-throughput single-cell isoform sequencing [4] |
RNA Sequencing Applications in Drug Discovery Pipeline
The integration of both short-read and long-read RNA sequencing technologies provides complementary strengths throughout the drug discovery pipeline. Short-read sequencing remains the workhorse for large-scale gene expression profiling, particularly in early screening stages where cost-effectiveness and high throughput are paramount. Its established protocols and extensive analytical frameworks make it suitable for applications where gene-level expression provides sufficient information.
Long-read sequencing technologies have matured to offer highly comparable gene-level quantification while providing the additional dimension of isoform-resolution data [16]. The ability to directly sequence full-length transcripts enables researchers to identify novel isoforms, characterize fusion genes, detect allele-specific splicing, and investigate RNA modifications—all of which can provide crucial insights for drug discovery [6]. As these technologies continue to evolve, with increasing throughput and decreasing costs, they are positioned to become foundational tools for unraveling the complexity of the transcriptome in health and disease.
For drug discovery pipelines, the strategic integration of both technologies offers the most powerful approach: using short-read sequencing for large-scale screening and long-read sequencing for deep investigation of mechanisms, biomarkers, and resistance patterns. This multi-technology strategy will ultimately enhance the efficiency of drug development, improve success rates in clinical trials, and advance the era of precision medicine.
The field of RNA sequencing has expanded far beyond standard gene expression profiling, branching into specialized applications that provide deeper biological insights. The ongoing comparison between short-read and long-read sequencing technologies is particularly relevant in these specialized contexts, where each approach offers distinct advantages and limitations. Short-read sequencing, typically performed on Illumina platforms, provides high-throughput, high-accuracy data at the gene level, making it suitable for quantifying expression levels. In contrast, long-read technologies from Pacific Biosciences and Oxford Nanopore Technologies sequence full-length transcripts, preserving isoform information and enabling the detection of structural variations, fusion genes, and RNA modifications without inferential assembly. This guide objectively compares how these technological approaches perform across three critical specialized applications: spatial transcriptomics, fusion gene detection, and RNA modification analysis, providing researchers with experimental data and methodologies to inform their platform selection.
Spatial Transcriptomics: Capturing Gene Expression in Context
Spatial transcriptomics (ST) has emerged as a revolutionary technology that bridges single-cell RNA sequencing with tissue architecture, enabling researchers to analyze gene expression patterns within their native spatial context. This integration provides unprecedented insights into cellular states, intercellular interactions, and tissue organization, with particular significance for cancer biology, developmental biology, and neuroscience [54] [55].
Technology Platform Comparison
Recent systematic benchmarking studies have evaluated four commercially available high-throughput ST platforms with subcellular resolution: Stereo-seq v1.3, Visium HD FFPE, CosMx 6K, and Xenium 5K. These platforms represent diverse technological strategies with varying performance characteristics [54].
Table 1: Performance Comparison of Subcellular Spatial Transcriptomics Platforms
| Platform | Technology Type | Resolution | Gene Panel Size | Key Strengths | Sensitivity Observations |
|---|---|---|---|---|---|
| Stereo-seq v1.3 | Sequencing-based (sST) | 0.5 μm | Whole transcriptome (poly(dT) capture) | Unbiased whole-transcriptome analysis | High correlation with scRNA-seq |
| Visium HD FFPE | Sequencing-based (sST) | 2 μm | 18,085 genes | Optimized for FFPE samples | Outperformed Stereo-seq for cancer cell markers |
| CosMx 6K | Imaging-based (iST) | Single molecule | 6,175 genes | Single-molecule precision | Higher total transcripts but lower correlation with scRNA-seq |
| Xenium 5K | Imaging-based (iST) | Single molecule | 5,001 genes | Superior sensitivity for marker genes | Highest sensitivity among tested platforms |
Experimental Factors Influencing RNA Capture Efficiency
The performance of spatial transcriptomics platforms is significantly influenced by several experimental factors that affect RNA capture efficiency:
Tissue Processing Methods: Formalin-fixed paraffin-embedded (FFPE) samples, while clinically practical, present challenges for RNA capture due to nucleic acid cross-linking and fragmentation caused by formaldehyde fixation. Fresh-frozen tissues generally maintain higher RNA integrity but require stringent storage conditions [56].
Section Thickness and Permeation: Tissue section thickness is critical for optimal RNA capture. Thick sections are difficult to penetrate, leading to RNA loss, while thin sections complicate obtaining complete cells. Permeation time must be carefully controlled, as incomplete penetration prevents mRNA capture, while excessive permeation causes mRNA drift to adjacent capture sites [56].
Probe Design Strategy: Traditional poly(T)-primed strategies primarily target mRNA with poly(A) tails but cannot capture non-coding RNAs and perform poorly with degraded RNA from FFPE samples. Stereo-seq V2 addresses this by using random hexamer primers (6N) instead of poly(T) primers for unbiased whole transcriptome capture [56].
Innovative Solutions for Enhanced Capture
Several innovative technologies have been developed to address the challenge of low RNA capture efficiency in spatial transcriptomics:
Decoder-seq: Utilizes dendrimer DNA nanostructures to create high-density spatial barcode arrays on three-dimensional nanoscale substrates, increasing DNA probe density approximately tenfold and achieving a detection sensitivity of 40.1 mRNA molecules per μm² [56].
MAGIC-seq: Employs a grid-based microfluidic "splicing chip" design that enables high-throughput, wide-field spatial transcriptome analysis. This approach significantly reduces chip preparation costs to approximately $0.11/mm² while minimizing batch effects for large-scale studies [56].
FaST Pipeline: A computational solution for rapid analysis of subcellular resolution spatial transcriptomics datasets. This pipeline can process datasets containing >500 million reads in approximately one hour on a standard workstation, enabling RNA-based cell segmentation without requiring imaging data [57].
Fusion Gene Detection: Methodological Considerations
Gene fusions represent important oncogenic drivers in cancer, with rapid and accurate detection being crucial for clinical decision-making. RNA sequencing has proven particularly effective for fusion detection, though methodological variations significantly impact performance [58].
Whole Transcriptome Sequencing Assay Development
A recently developed whole transcriptome sequencing (WTS) assay for fusion gene detection demonstrates the critical parameters for optimal performance:
Table 2: Performance Characteristics of WTS Fusion Detection Assay
| Parameter | Threshold for Optimal Sensitivity | Impact on Detection |
|---|---|---|
| RNA Degradation (DV200) | ≥ 30% | Below this threshold, sensitivity decreases significantly |
| RNA Input | > 100 ng | Insufficient input reduces fusion detection capability |
| Fusion Expression | ≥ 40 copies/ng | Low expression fusions may be missed |
| Number of Mapped Reads | > 80 million reads | Lower coverage reduces detection sensitivity |
This optimized WTS assay successfully identified 62 out of 63 known gene fusions, achieving a sensitivity of 98.4% with 100% specificity in validation studies [58].
Short-read vs. Long-read Approaches for Fusion Detection
The Fuzzion2 algorithm represents a significant advancement for fusion detection using short-read RNA sequencing data. This method employs fuzzy pattern matching and frequency minimizers to analyze unmapped RNA-seq samples in minutes with accuracy exceeding current methods, using a reference of 21,736 patterns representing chimeric fusions and internal tandem duplications [59].
For long-read approaches, specialized library preparation methods like PacBio's MAS-ISO-seq (now relabeled as Kinnex full-length RNA sequencing) enable fusion detection by concatenating full-length transcripts into longer fragments (10-15 kb average size) that are sequenced and then bioinformatically decomposed to original transcripts. This approach preserves the complete structural information of fusion events [4].
Computational Considerations and Filtering Strategies
Fusion detection by RNA sequencing remains challenging with a high rate of false positives common across algorithms. Careful assessment of RNA quality and appropriate filtering strategies are required for reliable clinical application. The WTS assay developed by Shanghai Pulmonary Hospital implemented a reportable list of 553 genes (from approximately 22,000 mRNA-encoding genes) based on clinical relevance, significantly reducing false positives while maintaining sensitivity for clinically actionable fusions [58].
RNA Modification Analysis: Expanding the Epitranscriptome
RNA modifications represent a crucial layer of post-transcriptional gene regulation, with emerging roles in cancer development and progression. Different sequencing approaches offer distinct capabilities for detecting these modifications [60].
Analytical Approaches for Modification Detection
Long-read Direct RNA Sequencing: Oxford Nanopore Technologies' direct RNA sequencing has become a valuable method for studying RNA modifications such as N6-methyladenosine (m6A) and pseudouridine (pseU). Recent advancements in RNA004 chemistry substantially reduce sequencing errors compared to previous chemistries, promising enhanced accuracy for epitranscriptomic analysis [61].
Computational Tool Performance: Benchmarking studies of modification detection tools for RNA004 data reveal that Dorado demonstrates higher recall (~0.92) than m6Anet (~0.51) for m6A sites with ≥10% modification ratio and ≥10X coverage. However, both tools can have high per-site false discovery rates (~40% for Dorado and ~80% for m6Anet), highlighting the need for careful interpretation and validation [61].
Short-read Indirect Methods: Though not providing direct modification detection, short-read sequencing can infer modification status through specialized protocols like immunoprecipitation-based methods (e.g., MeRIP-seq) for modifications such as m6A.
RNA Modification Roles in Cancer
Comprehensive profiling of RNA modification-related genes across multiple cancer types (breast, colon, liver, and lung) has identified three candidate genes with increased expression in cancer tissues associated with poor survival: the 5-methylcytosine methyltransferases NSUN2 and DNMT3B, and CBP20, an N7-methylguanosine binding protein. Functional validation confirmed that CBP20 knockdown reduced cancer cell viability, induced apoptosis, and caused G1-S cell cycle arrest, establishing it as a potential therapeutic target [60].
Integrated Experimental Protocols
Spatial Transcriptomics Workflow with FaST Pipeline
The Fast analysis of Spatial Transcriptomics (FaST) pipeline provides an efficient workflow for analyzing subcellular resolution datasets [57]:
Flowcell Barcode Map Preparation: HDMI fastq files from the first sequencing round are processed to create a "flow cell barcode map" associating barcodes with x and y coordinates.
Sample Fastq Reads Preprocessing: R1 reads (containing spatial barcodes) are compared with the flowcell barcode map index to identify tiles used for RNA capture.
Reads Alignment: STAR aligner processes reads while preserving spatial coordinate information as BAM tags.
Digital Gene Expression: BAM files are split for parallel processing tile by tile, with custom Perl scripts parsing genomic intervals and assigning subcellular localizations.
RNA-based Cell Segmentation: Nuclear localized transcripts are used to generate putative nuclear masks, followed by segmentation using intron counts and entire read matrices.
Whole Transcriptome Sequencing for Fusion Detection
The validated WTS assay for fusion detection employs the following methodology [58]:
RNA Extraction and Quality Control: Total RNA is extracted from FFPE samples using RNeasy FFPE Kit, with assessment via NanoDrop 8000, Qubit 3.0, and Agilent 2100 Bioanalyzer. DV200 value ≥30% is required.
rRNA Depletion: Ribosomal RNA is removed using NEBNext rRNA Depletion Kit (Human/Mouse/Rat).
Library Preparation: Using NEBNext Ultra II Directional RNA Library Prep Kit with custom adaptor and index primers. Fragmentation is skipped for samples with DV200 ≤50%.
Sequencing: Performed on Gene+seq 2000 instrument, generating approximately 25 Gb of 100 bp paired-end reads per sample.
Bioinformatic Analysis: Implementation of filtering strategies based on reportable gene list and expression thresholds.
Essential Research Reagent Solutions
Table 3: Key Research Reagents for Specialized RNA Applications
| Reagent/Kit | Application | Function | Considerations |
|---|---|---|---|
| RNeasy FFPE Kit | Fusion detection (WTS) | RNA extraction from FFPE samples | Critical for obtaining quality RNA from archived clinical samples |
| NEBNext rRNA Depletion Kit | Fusion detection (WTS) | Removal of ribosomal RNA | Enhances sequencing coverage of mRNA targets |
| NEBNext Ultra II Directional RNA Library Prep Kit | Fusion detection (WTS) | Library preparation for RNA-seq | Maintains strand specificity for accurate fusion mapping |
| MAS-ISO-seq for 10x Genomics | Long-read scRNA-seq | Library prep for full-length transcript sequencing | Enables isoform and fusion detection in single cells |
| Chromium Single Cell 3' Reagent Kits | Spatial transcriptomics | Single-cell partitioning and barcoding | Generates full-length cDNA for downstream applications |
| Spateo-release Package | Spatial transcriptomics | RNA-based cell segmentation | Enables analysis without imaging data |
The selection between short-read and long-read RNA sequencing technologies for specialized applications depends on the specific research questions and experimental requirements. Spatial transcriptomics benefits from the high resolution offered by both sequencing-based (Stereo-seq, Visium HD) and imaging-based (Xenium, CosMx) platforms, with choice dependent on the need for whole transcriptome analysis versus higher sensitivity for targeted panels. Fusion gene detection achieves high sensitivity with optimized short-read WTS approaches, while long-read technologies provide unambiguous fusion transcript characterization. RNA modification analysis is particularly advanced by long-read direct RNA sequencing, though computational tools require careful validation due to substantial false discovery rates. As these technologies continue to evolve, multimodal approaches that leverage the complementary strengths of both short-read and long-read methodologies will likely provide the most comprehensive insights into RNA biology.
Navigating Practical Challenges: From Sample Prep to Data Analysis
The journey of RNA sequencing (RNA-Seq) begins with the critical step of library preparation, a process that converts RNA into a format compatible with high-throughput sequencing platforms. This complex workflow is a major source of technical variability that can significantly impact data quality and interpretation. Library preparation involves multiple sophisticated steps, including RNA extraction, fragmentation, adapter ligation, and amplification, each introducing potential biases that researchers must understand and control for [62] [63].
The fundamental goal of library preparation is to represent the original transcript population as faithfully as possible while incorporating necessary sequences for the sequencing process. However, the intricate nature of this process means that different approaches yield substantially different outcomes in terms of transcript coverage, detection of isoforms, and quantification accuracy [64]. As RNA-Seq continues to evolve as the gold standard for transcriptome analysis, recognizing how library preparation choices affect downstream results becomes paramount for generating biologically meaningful data, particularly in the context of comparing short-read and long-read sequencing technologies [30].
Comparative Workflows: Short-Read vs. Long-Read Library Preparation
Short-Read Library Preparation
Short-read RNA-Seq protocols, dominant in platforms like Illumina, involve a multi-step process that typically includes RNA fragmentation early in the workflow. The standard approach begins with RNA extraction and purification, followed by enrichment of desired RNA species through poly(A) selection or ribosomal RNA depletion [62] [30]. The RNA is then fragmented—either enzymatically or chemically—before being reverse-transcribed into cDNA. Adapters containing barcode sequences for multiplexing are ligated, and the library is amplified via PCR to generate sufficient material for sequencing [63].
A key characteristic of short-read preparation is the fragmentation-first approach, which breaks RNA into smaller pieces (typically 200-500 bp) before sequencing. This enables high sequencing accuracy but reconstructs transcript isoforms bioinformatically, which can be challenging for complex transcriptomes [30]. The multistep nature of this process introduces multiple potential bias sources, including fragmentation bias, random hexamer priming bias, adapter ligation bias, and PCR amplification bias [62].
Long-Read Library Preparation
Long-read technologies from PacBio and Oxford Nanopore Technologies (ONT) employ fundamentally different preparation strategies. PacBio's Single Molecule Real-Time (SMRT) sequencing typically uses a full-length cDNA approach, where reverse transcription produces complete cDNA copies of RNA molecules before any fragmentation occurs [10]. These full-length cDNAs are then converted into SMRTbell library constructs with hairpin adapters on both ends, creating circular templates that can be sequenced repeatedly to generate highly accurate HiFi reads [65].
ONT's nanopore sequencing offers direct RNA sequencing capabilities, potentially bypassing reverse transcription altogether, though cDNA-based approaches are also common. The defining feature is that RNA or cDNA molecules are sequenced in their entirety without fragmentation, preserving length information that is crucial for isoform identification [10]. This approach eliminates fragmentation biases and provides direct access to complete transcript sequences, though it has historically faced challenges with higher error rates that are now being addressed through improved chemistries and base-calling algorithms [10].
Table 1: Key Differences Between Short-Read and Long-Read Library Preparation
| Parameter | Short-Read cDNA-Seq | Long-Read cDNA-Seq | Long-Range RNA-Seq |
|---|---|---|---|
| Platform Examples | Illumina, Ion Torrent | PacBio | Oxford Nanopore |
| Typical Read Length | 50-300 bp | 1-50 kb | 1-50 kb |
| Fragmentation Approach | RNA fragmented before sequencing | cDNA fragmented after full-length synthesis | Minimal to no fragmentation |
| Amplification Requirements | PCR amplification typically required | PCR often required | Can be PCR-free |
| Key Advantages | High accuracy, cost-effective for high coverage | Full-length transcript capture simplifies isoform analysis | Direct RNA sequencing, detects modifications |
| Key Limitations | Limited isoform detection, assembly required | Lower throughput, higher input requirements | Higher error rates, specialized equipment |
Workflow Visualization
The following diagram illustrates the key differences in library preparation workflows between short-read and long-read sequencing approaches:
Diagram 1: Library Preparation Workflow Comparison
Technical biases in RNA-Seq library preparation arise from multiple steps in the workflow, potentially distorting the representation of true transcript abundances. Understanding these biases is essential for experimental design and data interpretation.
Fragmentation bias represents a fundamental difference between short-read and long-read approaches. Short-read protocols fragment RNA before sequencing, which can introduce sequence-specific cleavage preferences and under-represent transcripts with specific structural features [62]. Chemical fragmentation using zinc shows more random patterns compared to enzymatic methods like RNase III, which may not cleave completely randomly, reducing sequence complexity [62]. Long-read technologies largely avoid this bias by sequencing full-length transcripts without fragmentation.
Amplification bias remains a significant challenge, particularly for low-input samples. PCR amplification can stochastically introduce biases that propagate through later cycles, with different molecules having unequal amplification probabilities [62]. The number of PCR cycles, polymerase choice (e.g., Kapa HiFi versus Phusion), and template GC content all influence amplification efficiency. For extremely AT/GC-rich sequences, additives like TMAC or betaine can help, along with adjusted extension temperatures and denaturation times [62].
Primer-related biases affect both technologies but manifest differently. Short-read protocols commonly use random hexamers for reverse transcription, which can exhibit sequence-specific priming efficiency and mispriming artifacts [62]. Long-read approaches may incorporate primers during cDNA synthesis that similarly show sequence preferences. Some protocols attempt to circumvent this by directly ligating adapters to RNA fragments, bypassing random priming altogether [62].
Adapter ligation bias stems from the substrate preferences of enzymes like T4 RNA ligase, which may favor certain sequence motifs over others. This can be mitigated by using adapters with random nucleotides at ligation junctions [62]. Additionally, mRNA enrichment bias varies by method—poly(A) selection introduces 3'-end capture bias, while rRNA depletion better preserves coverage uniformity but may capture more non-coding RNA [62] [64].
Bias Distribution Across Library Preparation Steps
The following diagram illustrates where major biases occur throughout the library preparation workflow:
Diagram 2: Bias Sources in Library Preparation Workflow
Fragmentation Strategies and Their Applications
Fragmentation Methods in RNA-Seq
Fragmentation represents a critical divergence point between short-read and long-read approaches, with profound implications for data quality and applications. In short-read sequencing, fragmentation is essential to generate molecules of appropriate length for sequencing platforms, typically creating 200-500 base pair fragments [63]. The two primary methods—chemical and enzymatic fragmentation—exhibit different bias profiles that must be considered during experimental design.
Chemical fragmentation using divalent cations under elevated temperature (e.g., zinc-based fragmentation) generally produces more random cleavage patterns, making it preferable for quantitative applications [62]. Enzymatic methods using RNase III or other nucleases may show sequence or structure-specific preferences, potentially reducing library complexity [62]. Some protocols reverse the order by creating full-length cDNA first, then fragmenting DNA mechanically or enzymatically, which can provide different bias profiles [62].
Long-read approaches fundamentally differ by minimizing or eliminating fragmentation. PacBio's SMRT sequencing typically uses full-length cDNA synthesis followed by optional DNA fragmentation only if needed for size selection [10]. Oxford Nanopore's direct RNA sequencing requires no fragmentation at all, preserving the native RNA molecule integrity. This absence of fragmentation enables one of long-read sequencing's key advantages: direct observation of complete transcript isoforms without computational assembly [30] [10].
Impact of Fragmentation on Data Outcomes
The fragmentation approach directly influences multiple aspects of sequencing data. Short-read fragmentation creates uniform coverage across transcripts when random, but sequence-specific biases can generate coverage artifacts that complicate isoform quantification [62]. The position of reads relative to transcript features also varies—fragmentation-based methods typically distribute reads across the entire transcript, while 3'-end focused methods (like some multiplexing-optimized protocols) concentrate reads at transcript ends [66].
For degraded samples like FFPE tissues, the inherent RNA fragmentation complicates standard protocols. In these cases, fragmentation-free approaches often perform better since additional fragmentation would further reduce useful sequence length [67]. Recent kit comparisons for FFPE samples demonstrate that both Takara SMARTer and Illumina Stranded Total RNA kits can generate usable data from degraded samples, with the Takara kit showing particular advantage for low-input scenarios despite higher ribosomal RNA content [67].
Table 2: Fragmentation Methods and Their Characteristics
| Fragmentation Method | Typical Applications | Advantages | Limitations | Recommended Solutions |
|---|---|---|---|---|
| Chemical Fragmentation | Standard short-read RNA-Seq | More random cleavage, better coverage uniformity | Requires optimization of time/temperature conditions | Use zinc-based rather than metal ion methods for improved randomness |
| Enzymatic Fragmentation | High-throughput short-read sequencing | Faster, easier to automate | Potential sequence/structural biases | Use multiple enzymes or optimized mixes to reduce bias |
| cDNA Fragmentation | Long-read sequencing, some short-read protocols | Avoids RNA degradation issues, more stable template | Additional reverse transcription step required | Use mechanical shearing for most random distribution |
| No Fragmentation | Full-length isoform analysis, direct RNA sequencing | Preserves complete transcript information, no assembly required | Longer reads may have higher error rates | Use circular consensus sequencing (HiFi) for improved accuracy |
Multiplexing Strategies for Scalable Sequencing
Principles of Multiplexing in RNA-Seq
Multiplexing represents a crucial strategy for enhancing throughput and reducing costs in both short-read and long-read RNA-Seq. The fundamental concept involves pooling multiple individually barcoded libraries for simultaneous sequencing, followed by computational separation (demultiplexing) based on these barcodes [65]. This approach allows researchers to maximize sequencer capacity by distributing costs across multiple samples while minimizing technical batch effects through simultaneous processing.
The core requirement for effective multiplexing is a robust barcoding system comprising unique oligonucleotide sequences ligated to each sample during library preparation. Effective barcodes must be easily distinguishable even in the presence of sequencing errors, typically requiring careful design to ensure balanced GC content and sufficient sequence divergence [65]. Modern platforms support extensive barcode sets—PacBio offers 384 unique barcodes for their SMRTbell adapters, while Illumina's systems provide diverse indexing options compatible with high-level multiplexing [65].
A critical performance metric in multiplexed experiments is pooling uniformity, representing how evenly sequencing data distributes across samples. This is typically measured using the coefficient of variation (CV), calculated as the standard deviation divided by the mean of data yield across samples [65]. Low CV values indicate high uniformity, essential for comparative analyses like differential expression where uneven coverage could introduce technical artifacts.
Multiplexing Implementation Across Platforms
Implementation details differ significantly between short-read and long-read platforms. Short-read multiplexing typically occurs during library preparation through adapter ligation with index sequences, allowing dozens of samples to be pooled in a single lane [68] [63]. The extremely high throughput of short-read platforms (millions to billions of reads per run) makes them particularly suitable for large-scale studies requiring extensive multiplexing.
Long-read platforms have historically offered lower throughput, making multiplexing essential for cost-effective experimentation. PacBio's Revio system dramatically improves this capacity, generating 15 times more HiFi data than previous systems [65]. Their approach uses SMRTbell adapter indexes with unique barcodes flanking DNA inserts, enabling efficient pooling and demultiplexing [65]. For RNA-specific applications, Kinnex adapter indexes allow sample-level multiplexing while capturing full-length isoform information that short-read methods miss.
Oxford Nanopore's multiplexing strategies leverage barcoding during cDNA synthesis, similar to PacBio's approach. A key advantage of nanopore multiplexing is the real-time data generation, allowing researchers to stop sequencing once sufficient coverage is achieved for each sample, potentially optimizing run efficiency [10]. However, achieving uniform coverage across multiplexed samples remains more challenging with nanopore technology compared to Illumina platforms.
Experimental Data and Performance Comparisons
Library Preparation Kit Performance
Recent comparative studies provide valuable insights into how different library preparation strategies perform across various sample types and conditions. A 2025 study directly compared two FFPE-compatible stranded RNA-seq kits: TaKaRa SMARTer Stranded Total RNA-Seq Kit v2 (Kit A) and Illumina Stranded Total RNA Prep Ligation with Ribo-Zero Plus (Kit B) [67]. Both kits generated high-quality data from challenging FFPE samples, but with important distinctions. Kit A achieved comparable gene expression quantification to Kit B while requiring 20-fold less RNA input (a crucial advantage for limited samples), albeit with increased sequencing depth requirements and higher ribosomal RNA content (17.45% vs. 0.1%) [67].
A comprehensive 2019 evaluation of four RNA-Seq kits revealed distinct performance characteristics across multiple parameters [64]. The Illumina TruSeq Stranded mRNA kit demonstrated superior performance for standard protein-coding gene analysis, while the TruSeq Stranded Total RNA kit provided better coverage of non-coding RNAs. The study found that despite technical differences, all kits allowed identification of a similar set of differentially expressed genes, suggesting that biological signals remain detectable across preparation methods [64].
For low-input scenarios, the SMARTer Ultra Low RNA Kit showed particular utility, though it exhibited bias against transcripts with high GC content [64]. The modified NuGEN Ovation protocol tended to capture longer genes compared to Illumina kits, which preferentially enriched for genes with higher expression and GC content [64]. These findings highlight how kit selection should align with experimental priorities, as no single method excels across all applications.
Quantitative Comparison of Library Preparation Methods
Table 3: Performance Metrics Across Library Preparation Kits
| Kit/Platform | Input Requirements | rRNA Depletion Efficiency | Exonic Mapping Rate | 3'/5' Bias | Best Applications |
|---|---|---|---|---|---|
| Illumina TruSeq Stranded mRNA | 100 ng - 1 μg | High (polyA selection) | High (~90%) | Moderate | Standard gene expression, protein-coding focus |
| Illumina TruSeq Stranded Total RNA | 100 ng - 1 μg | Moderate (rRNA depletion) | Moderate-High | Low | Whole transcriptome, inc. non-coding RNA |
| Takara SMARTer Stranded Total RNA | 1 ng - 10 ng | Moderate | Moderate | Variable | Low input, degraded samples |
| PacBio Kinnex RNA | Varies by application | Variable | High for isoforms | Minimal | Full-length isoform analysis, fusion detection |
| Oxford Nanopore Direct RNA | No cDNA conversion needed | Not applicable | Reference-dependent | Minimal | RNA modification detection, real-time analysis |
Impact on Differential Expression Analysis
The choice of library preparation method directly influences power for differential expression detection. Studies examining experimental design parameters have demonstrated that biological replication provides substantially more power than technical replication or increased sequencing depth [68]. In fact, sequencing depth could be reduced to as low as 15% without substantial impacts on false positive or true positive rates when adequate biological replicates were included [68].
Multiplexing strategies directly enable this improved experimental design by making larger sample sizes economically feasible. With fixed budgets, researchers often face the trade-off between sequencing depth and sample size. The evidence strongly supports prioritizing sample size over depth for differential expression studies, as the statistical power gained from additional biological replicates outweighs the benefits of deeper sequencing [68]. This makes efficient multiplexing strategies essential for robust experimental design.
Different analysis tools also show varying sensitivity to library preparation artifacts. In comparative evaluations, the DESeq algorithm performed more conservatively than edgeR and NBPSeq, though all three methods based on negative binomial distributions showed generally concordant results [68]. These tools have evolved to accommodate technical artifacts, but library preparation choices still influence their performance, particularly for low-abundance transcripts or subtle expression differences.
Research Reagent Solutions for Library Preparation
Successful RNA-Seq library preparation requires careful selection of reagents and kits tailored to specific research needs. The following table outlines key solutions across different applications:
Table 4: Essential Research Reagents for RNA-Seq Library Preparation
| Reagent/Kit | Primary Function | Key Features | Best Suited Applications |
|---|---|---|---|
| Illumina Stranded Total RNA Prep with Ribo-Zero Plus | rRNA depletion | Effective rRNA removal (~99.9%), preserves strand information | Whole transcriptome analysis including non-coding RNA |
| Takara SMARTer Stranded Total RNA-Seq Kit v2 | Low-input library prep | Requires only 1 ng input, maintains strand specificity | Limited samples, small biopsies, rare cell populations |
| PacBio SMRTbell Adapter Indexes | Long-read multiplexing | 384 unique barcodes, high demultiplexing accuracy | Full-length isoform analysis, population-scale studies |
| Kinnex Adapter Indexes | RNA multiplexing | Compatible with full-length cDNA synthesis, same barcodes across kits | Bulk and single-cell RNA-seq on PacBio platforms |
| Twist Bioscience UDI Adapters | Ultra-low input multiplexing | Unique dual indexes reduce cross-talk, compatible with Ampli-Fi | Low DNA input applications, single-cell genomics |
| Kapa HiFi Polymerase | Library amplification | High fidelity, reduced GC bias, improved uniformity | All applications requiring PCR amplification |
Library preparation complexities remain significant determinants of RNA-Seq outcomes, with fragmentation strategies, bias mitigation, and multiplexing approaches differentiating short-read and long-read technologies. The experimental evidence demonstrates that each method carries distinct advantages—short-read protocols offer established, cost-effective solutions for standard gene expression analysis, while long-read technologies excel in isoform resolution and structural variant detection without assembly.
The future of RNA-Seq library preparation lies in addressing current limitations while expanding applications. For short-read sequencing, reducing amplification requirements and improving coverage uniformity remain active development areas. For long-read technologies, increasing throughput and reducing costs while maintaining accuracy are key priorities. Emerging solutions like PCR-free protocols, isothermal amplification, and hybrid capture methods continue to evolve, offering researchers an expanding toolkit for transcriptome analysis [62] [65].
As multiplexing capabilities advance on both short-read and long-read platforms, researchers gain increasing flexibility in experimental design, enabling more sophisticated studies with appropriate biological replication. The integration of molecular barcoding and unique dual indexes further enhances accuracy by enabling the identification of PCR duplicates. By understanding the complexities, biases, and strategic considerations outlined in this guide, researchers can make informed decisions that optimize their RNA-Seq experiments for specific research questions and sample types.
RNA quality is a pivotal factor in the success of any RNA sequencing study, directly influencing the accuracy and reliability of gene expression quantification. The RNA Integrity Number (RIN) has emerged as the standard metric for assessing RNA quality, with scores ranging from 10 (perfect) to 1 (completely degraded) [69]. While short-read sequencing has traditionally demonstrated greater tolerance for partially degraded RNA, recent advances in long-read technologies from PacBio and Oxford Nanopore Technologies (ONT) are redefining their applicability to a broader range of sample qualities [70] [6]. This guide provides an objective comparison of how these sequencing approaches perform with degraded and challenging samples, supported by experimental data, to help researchers select the appropriate technology for their specific sample quality constraints.
The fundamental challenge with degraded RNA lies in its non-uniform effect across transcripts. Unlike regulated biological decay in living cells, post-mortem or ex vivo degradation often occurs stochastically, though evidence suggests some transcript-specific patterns remain [69]. This degradation introduces systematic biases that can confound biological interpretations if not properly addressed. While samples with RIN > 8 are universally considered ideal, valuable research opportunities often involve samples with lower RIN values—particularly in clinical, field ecology, and biobank settings where immediate optimal preservation is challenging [70] [69].
Technology Comparison: Performance Across Sample Qualities
Short-Read Sequencing with Degraded RNA
Short-read RNA sequencing (primarily Illumina-based) has historically been the preferred choice for partially degraded samples due to its ability to sequence fragmented RNA. The technology leverages random priming during cDNA synthesis, enabling the capture of information from RNA fragments as small as 50-100 nucleotides [69]. This characteristic makes it particularly suitable for formalin-fixed paraffin-embedded (FFPE) samples and other challenging specimen types where fragmentation is inevitable.
Experimental data from degradation time-course studies demonstrate that short-read sequencing maintains robust gene-level quantification even with moderate degradation (RIN 5-7), though with some limitations. As RIN decreases, there is a predictable reduction in library complexity and a shift in read distribution toward the 3' end of transcripts [69]. One study systematically evaluating RNA degradation found that while principal component analysis clearly separated samples by RIN value, biological signals remained detectable even in substantially degraded samples (RIN ~4) when appropriate statistical correction was applied [69].
Table 1: Short-Read Sequencing Performance Across RNA Quality Levels
| RIN Range | Expected Mapping Rate | Key Limitations | Recommended Applications |
|---|---|---|---|
| 10-9 (Excellent) | 70-90% [71] | Minimal limitations | All applications, including alternative splicing analysis |
| 8-7 (Good) | 65-85% | Reduced detection of 5' ends | Gene-level differential expression |
| 6-5 (Moderate) | 60-75% | 3' bias in read distribution | Gene-level detection in valuable samples |
| <5 (Low) | 45-60% | Significant loss of library complexity | Exploratory analysis of unique samples |
Long-Read Sequencing with Degraded RNA
Long-read technologies face distinct challenges with degraded samples because they ideally require full-length transcripts for comprehensive isoform characterization. However, recent systematic benchmarks reveal nuanced performance characteristics across different long-read platforms and protocols.
Oxford Nanopore Direct RNA Sequencing (DRS) is particularly sensitive to RNA degradation because it sequences native RNA through a nanopore, requiring intact transcripts with preserved polyA tails for adapter ligation [70]. Degradation studies show that as RIN decreases, DRS data exhibit reduced library complexity, shorter read lengths, and an overrepresentation of shorter genes and isoforms [70]. Despite these limitations, research indicates that most genes and isoforms remain detectable even in degraded samples (RIN >7), and explicit correction for RNA integrity in differential expression analysis can recover meaningful biological signals [70].
PCR-cDNA Nanopore sequencing and PacBio Iso-Seq demonstrate greater resilience to moderate degradation through their amplification steps, which can rescue information from partially fragmented transcripts. The SG-NEx project, a comprehensive benchmark comparing RNA-seq protocols, found that PCR-amplified cDNA protocols consistently generated the highest throughput per sample and showed more uniform coverage across transcript lengths compared to direct RNA methods [6]. However, these protocols introduce their own biases, with transcripts from highly expressed genes being overrepresented in PCR-based approaches [6].
Table 2: Long-Read Technology Performance with Varying RNA Quality
| Technology/Protocol | Optimal RIN | Degraded Sample Tolerance | Key Degradation Effects |
|---|---|---|---|
| ONT Direct RNA | >9.5 [70] | Limited (RIN >7 with correction) | Strong 3' bias, reduced read length |
| ONT PCR-cDNA | 8-10 | Moderate (RIN >6) | Overrepresentation of highly expressed genes |
| PacBio Iso-Seq | 8-10 | Moderate (RIN >6) | Depletion of shorter transcripts |
| MAS-ISO-Seq (PacBio) | 8-10 | Moderate | Better retention of transcripts <500 bp |
Experimental Designs for Assessing Degradation Effects
Controlled Degradation Time-Series Experiments
Rigorous assessment of sequencing technology performance across RNA quality levels requires controlled degradation experiments. One robust approach involves collecting cell pellets and subjecting them to repeated freeze-thaw cycles at specific time intervals (e.g., 0, 0.5, 1, 2, 3, 4, 6, and 8 hours) before RNA extraction [70]. This method generates a series of samples with RIN values spanning from approximately 10 down to 7, creating a calibrated degradation gradient while preserving RNA yield and purity.
The experimental workflow typically follows: cell culture and harvesting → controlled degradation timeline → RNA extraction and quality assessment → library preparation with multiple technologies → sequencing and comparative analysis [70]. This design enables direct comparison of the same biological material across different degradation states and sequencing technologies, controlling for biological variability that could confound comparisons.
For such experiments, key quality control metrics include:
- RIN values for each time point
- RNA concentration and purity (A260/280 ratios)
- Library complexity metrics from sequencing data
- Mapping rates and coverage uniformity
- Transcript detection rates compared to undegraded controls
Spike-In Controls for Degradation Monitoring
Incorporating synthetic RNA spike-ins with known concentrations provides an internal standard for quantifying degradation effects on expression measurements. The SG-NEx project utilized multiple spike-in types including Sequins, ERCC, and SIRV variants to evaluate quantification accuracy across protocols [6]. These controls enable researchers to distinguish technical effects of degradation from biological signals and to calibrate normalization methods specifically for degraded samples.
Analysis Strategies for Degraded Samples
Computational Correction for RNA Quality
When working with degraded samples, specific analytical approaches can mitigate quality-related artifacts. For short-read data, tools like DegNorm implement degradation normalization by estimating gene-specific degradation rates from read coverage patterns [69]. The fundamental principle involves modeling the position-dependent bias in read distribution, which typically shows an exponential decrease from the 3' to 5' end of transcripts in degraded samples [72].
For experimental designs incorporating samples with varying RIN values, explicitly including RIN as a covariate in linear models for differential expression analysis can effectively remove degradation-related artifacts [69]. This approach has been shown to recover biological signals that would otherwise be confounded by RNA quality differences, particularly when the effect of interest is not correlated with RIN values.
Quality Control and Filtering Recommendations
Robust quality assessment is particularly critical when working with challenging samples. The nf-core/nanoseq pipeline provides a community-curated framework for long-read data, performing comprehensive quality control, alignment, and transcript quantification [6]. Key degradation-specific QC metrics include:
- Read length distribution shifts toward shorter fragments
- Coverage uniformity across transcripts
- End bias quantification (3'/5' ratios)
- Spike-in recovery rates for degraded samples
- Library complexity estimates via duplicate rates
Diagram: Sample quality decision framework for RNA sequencing. RIN assessment guides technology selection and analysis approach.
Table 3: Key Research Reagent Solutions for Degraded Sample Sequencing
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| RNeasy Lipid Tissue Kit (QIAGEN) | RNA extraction from challenging samples | Effective for degraded materials; used in systematic degradation studies [70] |
| NEXTflex polyA+ Beads (Bioo Scientific) | mRNA enrichment for long-read sequencing | PolyA selection requires minimally degraded RNA for optimal results [70] |
| Spike-in RNA Variants (SIRV, ERCC, Sequin) | Internal controls for quantification | Essential for evaluating technical performance in degradation experiments [6] |
| MAS-ISO-seq for 10x Genomics (PacBio) | Single-cell long-read library prep | Enables retention of shorter transcripts and removal of truncated cDNA artefacts [4] |
| NuGEN Ovation RNA-seq System | RNA amplification for low-input samples | Can improve library complexity from degraded samples [73] |
The choice between short-read and long-read technologies for degraded samples involves careful consideration of research priorities and sample limitations. Short-read sequencing remains the most robust choice for significantly degraded samples (RIN <6) when gene-level expression data is the primary goal. Its ability to utilize fragmented RNA provides more comprehensive transcript detection despite the loss of isoform-level information [69].
Long-read sequencing technologies have made significant strides in degraded sample tolerance, particularly through PCR-cDNA approaches that can effectively handle moderate degradation (RIN 6-8) [6]. When isoform discovery and characterization are paramount, and samples exhibit only moderate degradation, long-read methods can provide valuable insights not accessible through short-read approaches.
For the most challenging samples, including those with very low RIN values or where both gene-level quantification and isoform information are needed, a hybrid approach combining both technologies may offer the optimal solution, leveraging the strengths of each method to overcome their respective limitations in the face of RNA degradation.
For researchers embarking on transcriptomic studies, one of the most fundamental strategic decisions is selecting an appropriate sequencing technology. The choice largely centers on the trade-offs between the established, high-throughput capabilities of short-read sequencing and the superior resolution for complex genomic regions offered by emerging long-read technologies [74] [75]. Short-read sequencing, exemplified by Illumina platforms, delivers high data volume and accuracy at a lower cost per base, making it a robust tool for large-scale studies focused on variant detection and gene-level expression quantification [7] [75]. In contrast, long-read sequencing from PacBio and Oxford Nanopore Technologies (ONT) generates reads spanning thousands of bases, enabling the direct observation of full-length transcript isoforms, structural variants, and repetitive elements without the need for assembly [8] [74]. This guide provides an objective, data-driven comparison to help researchers balance throughput, coverage, and budget effectively.
Quantitative Technology Comparison
The following tables summarize the core performance metrics and application strengths of each technology, based on recent experimental benchmarks and market data.
Table 1: Key Performance Metrics for Short-Read and Long-Read Sequencing
| Metric | Short-Read Sequencing | Long-Read Sequencing |
|---|---|---|
| Typical Read Length | 50-300 base pairs [75] | Thousands to hundreds of thousands of base pairs [75] |
| Sequencing Accuracy | >99.9% [7] [75] | >99% (with recent HiFi reads) [74] [75] |
| Typical Application Depth | 5M to ≥25M reads per sample for gene expression [76] | Varies by application; lower throughput but higher clarity per molecule [74] |
| Relative Cost per Sample | More cost-effective for high-throughput [76] [74] | Higher cost per base; requires focus on cost per resolved question [74] |
| DNA/RNA Input Quality | Standard quality requirements | Requires high molecular weight DNA or high-quality RNA [75] |
Table 2: Application-Based Strengths and Limitations
| Application | Short-Read Performance | Long-Read Performance |
|---|---|---|
| Gene Expression Quantification | Excellent; high correlation between technical replicates [4] [77] | Excellent for gene-level; robust estimates from PCR-cDNA and IsoSeq [78] |
| Variant Detection (SNPs, Indels) | High accuracy and sensitivity [7] [79] | Effective, but performance varies by platform and coverage [7] |
| Transcript Isoform Detection | Limited; cannot reliably resolve full-length isoforms [8] [79] | Exceptional; enables end-to-end sequencing of full-length transcripts [8] [78] |
| Structural Variant Detection | Limited in complex/repetitive regions [74] [7] | Superior; resolves large insertions, deletions, and rearrangements [74] [7] |
| De Novo Genome Assembly | Challenging due to read length [75] | Highly effective for generating contiguous assemblies [75] |
Experimental Comparisons and Benchmarking Data
Protocol Comparison in Single-Cell and Bulk RNA Sequencing
A rigorous 2025 study sequenced the same 10x Genomics 3' cDNA from patient-derived organoid cells using both Illumina (short-read) and PacBio Sequel IIe (long-read) platforms. The researchers performed a per-molecule comparison by matching cell barcodes and unique molecular identifiers (UMIs). They found that both methods recovered a large proportion of cells and transcripts and yielded highly comparable gene expression results for relevant cancer genes [4].
However, platform-dependent biases were evident. Short-read sequencing provided a higher sequencing depth, while long-read sequencing (using MAS-ISO-seq library prep) allowed for the retention of transcripts shorter than 500 bp and the removal of a significant proportion of truncated cDNA contaminated by template switching oligos (TSO). The filtering of such artefacts, identifiable only from full-length transcripts, was noted as a factor that reduces gene count correlation between the two methods [4].
The Singapore Nanopore Expression (SG-NEx) project conducted a systematic benchmark of five RNA-seq protocols, including short-read cDNA, Nanopore direct RNA, direct cDNA, PCR-amplified cDNA, and PacBio IsoSeq. Among long-read protocols, PCR-amplified cDNA sequencing generated the highest throughput, with the most recent data matching short-read RNA-seq capacity. PacBio IsoSeq generated the longest reads on average, while PCR-amplified cDNA and IsoSeq data showed the most uniform coverage across transcript length and the highest proportion of reads spanning all exon junctions ("full-splice-match" reads) [78].
Variant Calling and Structural Variant Analysis in Cancer Genomics
A methodological comparison on colorectal cancer samples evaluated short-read Illumina and long-read Nanopore technologies for variant calling. The study reported that Illumina sequencing achieved a average mapping quality of 33.67 (99.96% accuracy), compared to Nanopore's average mapping quality of 29.8 (99.89% accuracy) [7].
While Illumina provided higher depth over target regions (e.g., ~105X for cancer samples versus ~21X for Nanopore), structural variant (SV) analysis revealed Nanopore's enhanced ability to resolve large and complex rearrangements, with consistently high precision across different SV types [7]. This underscores the complementary nature of these technologies: short-reads offer high accuracy and depth for small variants, while long-reads excel at resolving larger-scale genomic alterations.
Cost and Operational Considerations
Direct Cost Breakdown
The most expensive step in an RNA-seq experiment is often library preparation, though costs can vary significantly based on the protocol [76].
Table 3: Sample Cost Breakdown for mRNA-seq (using Illumina NovaSeq S4 flow cell at full capacity) [76]
| Cost Component | Illumina TruSeq (≥25M reads) | NEBnext Ultra II (20M reads) | 3' mRNA-seq (e.g., BRB-seq; 5M reads) |
|---|---|---|---|
| Library Prep | $68.7 | $41.3 | $24.0 |
| Sequencing | $36.9 | $25.9 | $4.6 |
| Data Analysis | ~$2.0 | ~$2.0 | ~$2.0 |
| Total per Sample | ~$113.9 | ~$75.5 | ~$36.9 |
It is critical to note that long-read sequencing carries a higher per-base cost, though this must be evaluated against the "cost per resolved biological question" [74]. For applications where isoform resolution or structural variant detection is the primary goal, long-read sequencing may provide greater value by delivering unambiguous answers that short-reads can only infer.
Strategic Selection Workflow
The following diagram outlines a decision-making workflow to guide researchers in selecting the most appropriate technology based on their project goals and constraints.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents and materials commonly used in sequencing experiments, as derived from the methodologies cited in this guide.
Table 4: Key Research Reagent Solutions for RNA Sequencing
| Item | Function/Description | Example Use Cases |
|---|---|---|
| 10x Genomics 3' Reagent Kits | Partitions cells into GEMs for single-cell RNA-seq; barcodes cDNA from individual cells. | Single-cell RNA sequencing of patient-derived organoids [4]. |
| MAS-ISO-seq Kit (PacBio) | Prepares libraries for long-read scRNA-seq; removes TSO artefacts and concatenates transcripts. | High-throughput full-length single-cell isoform sequencing [4]. |
| TruSeq Stranded mRNA Prep (Illumina) | Prepares stranded mRNA-seq libraries; preserves strand orientation during cDNA synthesis. | Standard short-read transcriptome profiling with strand information [77]. |
| BRB-seq Library Prep Kit | Early barcoding and pooling of samples for 3' mRNA-seq; drastically reduces library prep cost. | Ultra-affordable high-throughput transcriptomics for large sample cohorts [76]. |
| Poly-A Selection Beads | Purifies polyadenylated mRNA from total RNA by binding to oligo-dT sequences. | mRNA enrichment for most standard RNA-seq protocols [77]. |
| SPRI Beads | Solid-phase reversible immobilization beads for DNA size selection and clean-up. | Post-amplification cDNA purification and size selection in library prep [4]. |
| Spike-in RNA Controls | RNA molecules with known sequence and concentration added to samples for normalization. | Evaluating quantification accuracy and technical performance across protocols [78]. |
The decision between short-read and long-read sequencing is not a matter of identifying a superior technology, but rather of matching the tool to the specific biological question and experimental constraints. Short-read sequencing remains the most cost-effective solution for large-scale studies focused on gene expression quantification and small variant detection, offering high accuracy and throughput [76] [80]. Long-read sequencing is transformative for applications requiring resolution of transcript isoforms, structural variants, or complex genomic regions, providing biological insights that are simply not accessible with short-reads alone [8] [74] [78].
A growing and powerful strategy is the use of a hybrid approach, leveraging short-reads for high-depth quantification across many samples and long-reads for full-length structure determination on a subset of samples [74] [79]. This leverages the complementary strengths of both technologies to provide a more comprehensive view of the transcriptome while optimizing budgetary resources. As both technologies continue to evolve, with short-reads becoming more efficient and long-reads becoming more accurate and affordable, this integrated approach is poised to become the new standard in genomic research.
The fundamental difference between short-read and long-read RNA sequencing technologies dictates distinct computational strategies for data processing and analysis. While short-read sequencing, championed by platforms like Illumina, breaks transcripts down for high-throughput, gene-level analysis, long-read sequencing, offered by PacBio and Oxford Nanopore Technologies (ONT), sequences full-length transcripts in a single pass, providing immediate isoform-resolution data [4] [9]. This dichotomy extends throughout the entire data lifecycle, from the initial base calling and quality control to final transcript quantification and visualization. The choice of technology consequently imposes specific requirements on workflow design, software selection, and computational infrastructure. This guide objectively compares the data handling pipelines for both approaches, drawing on recent experimental benchmarks to outline their performance, strengths, and resource demands.
Workflow Architecture and Logical Pathways
The data analysis pipelines for short-read and long-read RNA sequencing, while sharing a common overarching goal of quantifying gene expression, diverge significantly in their specific steps and the types of artefacts they must address. The following diagram maps the logical flow and key decision points for each workflow.
Diagram 1: A logical workflow for RNA-seq data analysis. The pipeline splits into platform-specific steps after basecalling, with long-read sequencing requiring specialized tools for isoform resolution.
Key Computational Phases and Tool Comparison
The processing of RNA-seq data can be segmented into distinct computational phases, each requiring specialized software tools tailored to the characteristics of short or long reads.
Table 1: Core Bioinformatics Tools for Short-Read vs. Long-Read RNA Sequencing
| Analysis Phase | Short-Read Tools | Long-Read Tools | Key Functional Differences |
|---|---|---|---|
| Quality Control | FastQC, MultiQC | LongQC, NanoPack [81] | Long-read tools assess read length distribution and identify concatemers, which are specific to technologies like PacBio MAS-ISO-seq. |
| Read Alignment | STAR, HISAT2 | minimap2 [81] | minimap2 is optimized for long, error-prone reads and can efficiently map across splice junctions without a prior genome annotation. |
| Quantification | featureCounts, HTSeq | Salmon, StringTie2 | Long-read quantification tools leverage full-length transcript alignments to resolve isoform-level expression, moving beyond gene-level counts. |
| Isoform Analysis | Cufflinks, StringTie | SQANTI3 [4] [81] | SQANTI3 provides rigorous quality control and classification for identified isoforms, filtering artefacts like truncated cDNA. |
| Single-Cell Analysis | Cell Ranger, Seurat [82] | WF-Single-Cell (ONT) [4] | Long-read single-cell pipelines (e.g., for 10x data) generate isoform-level count matrices, enabling cell-atlas construction with isoform diversity. |
Specialized Toolkits for Single-Cell RNA Sequencing
Single-cell RNA sequencing (scRNA-seq) adds a layer of complexity, necessitating tools that can handle cell barcoding, unique molecular identifiers (UMIs), and significant technical noise. The following table details essential toolkits that form the backbone of modern single-cell analysis, applicable to both short-read and, increasingly, long-read data.
Table 2: Essential Bioinformatics Tools for Single-Cell RNA-Seq Analysis in 2025 [82]
| Tool | Function | Key Application in 2025 |
|---|---|---|
| Cell Ranger | Preprocessing of 10x Genomics data | Remains the gold standard for demultiplexing raw FASTQ files into gene-barcode count matrices; supports multiome (RNA+ATAC) data. |
| Seurat | Comprehensive scRNA-seq analysis in R | The most mature R toolkit for data integration, clustering, and visualization; natively supports spatial transcriptomics and multi-modal data. |
| Scanpy | Comprehensive scRNA-seq analysis in Python | Dominates large-scale analysis (millions of cells); integrates with the scverse ecosystem (e.g., scvi-tools, Squidpy) for advanced modeling. |
| scvi-tools | Deep generative modeling | Uses variational autoencoders for superior batch correction, imputation, and annotation; supports multiple modalities (RNA, ATAC, spatial). |
| CellBender | Ambient RNA removal | Employs deep learning to distinguish real cell signals from background noise, crucially improving cell calling and clustering in droplet-based data. |
| Harmony | Batch effect correction | Scalable algorithm that efficiently integrates datasets from different batches or donors while preserving biological variation. |
| Squidpy | Spatial transcriptomics analysis | Enables analysis of spatial neighborhood graphs, ligand-receptor interactions, and spatial clustering for platforms like 10x Visium and MERFISH. |
Experimental Data and Performance Benchmarks
Recent systematic studies provide quantitative data on the performance of short-read and long-read sequencing technologies, offering critical insights for platform selection.
Protocol Comparison from the SG-NEx Project
A landmark 2025 study from the Singapore Nanopore Expression (SG-NEx) project benchmarked five RNA-seq protocols across seven human cell lines. The core experimental protocol involved sequencing each cell line with multiple replicates using:
- Illumina short-read cDNA sequencing
- Nanopore direct RNA sequencing
- Nanopore amplification-free direct cDNA sequencing
- Nanopore PCR-amplified cDNA sequencing
- PacBio IsoSeq [6]
The resulting data allowed for a direct comparison of throughput, coverage, and accuracy, summarized in the table below.
Table 3: Experimental Performance Metrics of RNA-Sequencing Protocols [6]
| Sequencing Protocol | Average Throughput | Relative Read Length | 5'/3' Coverage Bias | Strength in Transcript Quantification |
|---|---|---|---|---|
| Illumina Short-Read | High | Short | High (due to fragmentation) | Robust gene-level expression |
| PacBio IsoSeq | Moderate | Longest | Uniform | Identification of major isoforms |
| ONT PCR-cDNA | Highest (for long-read) | Long | Uniform | High transcript coverage, full-splice-match reads |
| ONT Direct cDNA | Moderate | Long | Uniform | Avoids PCR amplification biases |
| ONT Direct RNA | Lower | Long | Higher at 3' end | Direct RNA modification detection |
Key findings from this benchmark include:
- Gene Expression Correlation: Gene expression estimates from long-read data, particularly Nanopore PCR-cDNA and direct cDNA protocols, showed high correlation with short-read data and the lowest estimation error for spike-in RNAs [6].
- Coverage Uniformity: Long-read protocols, especially PCR-amplified cDNA and PacBio IsoSeq, demonstrated more uniform coverage across the length of transcripts compared to short-read data, which showed biases due to RNA fragmentation [6].
- Transcript Diversity: The PCR-amplified cDNA protocol was found to be biased towards highly expressed genes, while PacBio IsoSeq showed a significant depletion of shorter transcripts, indicating that library preparation methods influence the recovered transcript diversity [6].
Single-Cell Isoform Comparison Study
A 2025 study directly compared short-read (Illumina) and long-read (PacBio MAS-ISO-seq) sequencing performed on the same 10x Genomics 3' cDNA libraries from patient-derived organoid cells. The experimental protocol was designed for a per-molecule comparison by matching reads through cell barcodes and UMIs [4].
Key Experimental Findings:
- Data Comparability: Both methods recovered a large proportion of cells and transcripts and rendered "highly comparable" results for gene expression, including for cancer-relevant genes [4].
- Platform-Specific Biases: Each method introduced distinct biases. Short-read sequencing provided higher sequencing depth and generally recovered more UMIs per cell. In contrast, long-read sequencing enabled the retention of transcripts shorter than 500 bp and allowed for the bioinformatic removal of a "large proportion of truncated cDNA contaminated by template switching oligos (TSO)" [4].
- Impact of Filtering: Stringent filtering of sequencing artefacts, which is only possible with full-length long reads (e.g., via the SQANTI3 tool), was noted to reduce the correlation of gene counts between the two platforms. This highlights a trade-off between data purity and cross-platform consistency [4].
Data Storage and Computational Infrastructure
The differences in data characteristics between short-read and long-read technologies have direct implications for storage and computational resource planning.
Data Volume and Complexity: While long-read sequencers can generate terabytes of data, a key consideration is the data type. Long-read data, particularly from PacBio HiFi and ONT, provides more biological information per read (e.g., full haplotype, isoform, methylation status), which can justify the storage cost [81] [83]. The trend toward multi-omics integration—combining genomic, transcriptomic, and epigenomic data—further increases storage and computational demands [83] [84].
Computational Workloads: Long-read analyses often require more memory (RAM) and processing time during the alignment and assembly phases due to the handling of longer, more complex sequences. However, continuous improvements in algorithms (e.g., minimap2) are mitigating these challenges [81] [10].
Infrastructure Solutions:
- Cloud Computing: Platforms like AWS, Google Cloud, and Azure offer scalable, cost-effective solutions for the variable and intensive workloads in genomics, providing compliance with security standards like HIPAA and GDPR [83].
- Workflow Management: Tools like Nextflow and Snakemake are critical for ensuring reproducible and scalable analysis pipelines. The nf-core community, for instance, offers curated pipelines such as
nf-core/nanoseqfor long-read RNA-seq data, which includes modules for quality control, alignment, and differential expression [6] [84]. - Containerization: Technologies like Docker and Singularity are integral for packaging tools and dependencies, guaranteeing portability and consistency across different computing environments [84].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key reagents and materials used in a typical experiment comparing short-read and long-read sequencing from the same cDNA library, as described in the experimental data section [4].
Table 4: Key Research Reagent Solutions for a Comparative Sequencing Study
| Item | Function in the Experiment |
|---|---|
| 10x Genomics Chromium Single Cell 3' Reagent Kits (v3.1 Chemistry Dual Index) | To generate single-cell, full-length cDNA libraries from cell suspensions, providing the common starting material for both sequencing platforms. |
| MyOne SILANE Dynabeads | For the solid-phase reversible immobilization (SPRI) cleanup and capture of amplified cDNA after the reverse transcription reaction in GEMs. |
| MAS-ISO-seq for 10x Genomics Single Cell 3' Kit (PacBio) | To prepare the cDNA for long-read sequencing on the PacBio platform; includes steps for TSO artefact removal and concatenation of transcripts. |
| SPRI Beads | Used for multiple cleanup and size-selection steps in both Illumina and PacBio library preparation protocols. |
| Agilent 4200 TapeStation High Sensitivity D5000 ScreenTape | For qualitative and quantitative analysis of the amplified cDNA and final sequencing libraries to ensure integrity and correct size distribution. |
| Qubit 1X dsDNA High Sensitivity Kit | For accurate quantification of DNA concentration in cDNA and final library preparations, crucial for loading the correct amount onto sequencers. |
| PacBio 3.2 Binding Kit | Used to prepare the SMRTbell polymerase complex for sequencing on the PacBio Sequel IIe system. |
| Illumina NovaSeq 6000 S/Xp/Xp4 Flow Cell | The consumable flow cell on which bridge amplification and short-read sequencing occur. |
| PacBio 8M SMRT Cell | The consumable cell containing the zero-mode waveguides for single-molecule, real-time (SMRT) long-read sequencing. |
The computational handling of RNA sequencing data is intrinsically linked to the choice of technology. Short-read workflows are mature, highly optimized for accurate gene-level quantification, and remain the gold standard for high-throughput applications like population-level transcriptomics. In contrast, long-read workflows are rapidly evolving to leverage their inherent advantage in resolving transcript isoforms, structural variations, and epigenetic modifications, albeit with different demands on data processing and storage [4] [6] [81].
The decision between them is not a matter of simple superiority but of strategic alignment with research goals. For gene-level differential expression in a large cohort, short-reads may be optimal. For uncovering isoform diversity, novel transcripts, or complex splicing in diseased tissues, long-reads are transformative [8]. The experimental evidence confirms that data from both methods can be highly comparable, yet the unique biases and filtering steps intrinsic to each platform lead to distinct outputs and biological insights. As both technologies continue to advance, the future of transcriptomics will likely see a more integrated approach, leveraging the complementary strengths of both short and long reads to build a complete and precise picture of gene expression.
The debate in genomics and transcriptomics often simplifies to a choice between short-read and long-read sequencing technologies. However, a new paradigm is emerging: hybrid and targeted approaches that combine their strengths to maximize biological insight. Short-read sequencing, known for its high throughput and accuracy, excels in quantifying gene expression and detecting small variants [4] [6]. In contrast, long-read sequencing provides full-length transcript information, enabling the precise characterization of isoform diversity, fusion transcripts, and complex genomic regions [6] [8]. By integrating these technologies or focusing them on specific genomic regions, researchers can overcome the inherent limitations of any single platform, offering a more comprehensive and accurate view of the genome and transcriptome. This guide compares these integrated strategies, providing experimental data and methodologies to inform their application in research and drug development.
Technical Comparison of Fundamental Technologies
Understanding the core characteristics of short-read and long-read sequencing is essential for appreciating how their combination creates a synergistic effect. The table below summarizes their key performance metrics.
Table 1: Core Technology Comparison of Short-Read and Long-Read RNA Sequencing
| Feature | Short-Read RNA-Seq | Long-Read RNA-Seq |
|---|---|---|
| Primary Strengths | High throughput, low cost per base, high per-base accuracy, established bioinformatics tools [6] [79] | Full-length transcript sequencing, isoform resolution, detection of novel transcripts & fusions, ability to detect RNA modifications [6] [8] |
| Typical Read Length | 50-300 bp [85] | Hundreds of bases to tens of kilobases [6] |
| Quantitative Gene Expression | Robust and established, lower estimation error for spike-ins in some benchmarks [6] | Highly comparable to short-read data, though platform-specific biases exist [4] [6] |
| Transcript Isoform Resolution | Limited; infers isoforms from fragmented reads, struggles with complex genes [6] | Excellent; directly sequences complete isoforms, even for complex genes [6] [79] |
| Coverage Uniformity | Biased due to RNA fragmentation; lower coverage at transcript ends [6] | More uniform; PCR-amplified cDNA and PacBio IsoSeq show superior coverage across transcripts [6] |
| Challenging Genomic Regions | Limited in repetitive regions, segmental duplications, and for phasing haplotypes [86] [79] | Superior in repetitive regions, segmental duplications, and for phasing variants [86] [79] |
Hybrid Sequencing: Integrating Broad and Long-Read Views
Hybrid sequencing leverages the high accuracy and coverage of short reads with the long-range information of long reads to create a more complete and accurate genomic picture. This approach is particularly powerful for building high-quality genome assemblies, phasing haplotypes, and comprehensively profiling complex transcriptomes.
Key Experimental Findings from Hybrid Approaches
- Expanded Benchmarking: The Genome in a Bottle Consortium (GIAB) used accurate long and linked reads to expand their benchmark variants for human genomes. This new benchmark added over 300,000 single nucleotide variants (SNVs) and 50,000 insertions or deletions (indels), covering 92% of the GRCh38 assembly and including many challenging, clinically relevant genes like PMS2 that were previously excluded [86].
- Transcriptome Complexity Resolution: The Singapore Nanopore Expression (SG-NEx) project conducted a systematic benchmark of five RNA-seq protocols. They found that while short-read RNA-seq provides robust gene expression estimates, long-read sequencing "more robustly identifies major isoforms" and facilitates the analysis of "alternative isoforms, novel transcripts, fusion transcripts and N6-methyladenosine RNA modifications" [6].
- Single-Cell Concordance: A study sequencing the same 10x Genomics 3' cDNA with both Illumina and PacBio platforms found that "both methods render highly comparable results and recover a large proportion of cells and transcripts." However, each method introduced platform-specific biases, such as long-read sequencing's ability to retain shorter transcripts and remove artifacts from truncated cDNA [4].
Representative Experimental Protocol: Comprehensive Transcriptome Profiling
The following workflow, based on the SG-NEx project, outlines a robust method for hybrid transcriptome sequencing [6]:
- Sample Preparation: Begin with high-quality RNA from the target cell lines or tissues.
- Multi-Protocol Library Construction: a. Short-read cDNA: Prepare libraries using a standard Illumina kit (e.g., poly(A) selection or rRNA depletion). b. Long-read Direct RNA: Prepare libraries for Nanopore sequencing without amplification to allow for native RNA modification detection. c. Long-read cDNA: Prepare PCR-amplified cDNA libraries for high-throughput Nanopore sequencing and/or PacBio IsoSeq libraries for long reads.
- Sequencing: Sequence short-read libraries on an Illumina platform (e.g., NovaSeq 6000) to a depth of 20-40 million reads per sample. Sequence long-read libraries on a PacBio or Nanopore platform to achieve high transcript coverage.
- Data Integration & Analysis: Use a standardized pipeline like nf-core/nanoseq for quality control, alignment, transcript discovery/quantification, and differential expression analysis. Integrate short-read data for high-confidence variant calling and long-read data for isoform identification and structural variant detection [6].
Targeted Sequencing: Focusing Power for Efficiency and Sensitivity
Targeted sequencing uses probes to enrich for specific genomic regions or transcripts of interest before sequencing. This approach significantly recreases the required sequencing depth and cost, while increasing sensitivity for detecting low-abundance variants or transcripts. It is especially valuable in clinical diagnostics where specific genes are of interest [87] [79].
Performance Comparison of Targeted Methods
Different probe-based methods offer distinct trade-offs between sensitivity and the ability to discover novel variants or fusion partners.
Table 2: Comparison of Targeted RNA-Seq Enrichment Methods
| Method | Mechanism | Key Strengths | Limitations | Supported Data |
|---|---|---|---|---|
| Amplicon-Based Multiplex PCR | Amplifies targets using multiple primer pairs [88] | Highest sensitivity; lowest limit of detection [88] | Limited ability to detect fusions with novel/uncommon partners [88] | Detects NTRK fusions with high clinical concordance [88] |
| Anchored Multiplex PCR | Amplifies sequences with one known gene-specific primer [88] | Detects fusions with unknown/novel partners [88] | - | Detects NTRK fusions with high clinical concordance [88] |
| Hybrid Capture-Based | Enriches targets using biotinylated probes [88] [87] | Detects fusions with unknown partners; suitable for cell-free DNA (cfDNA) [88] [87] | - | 93.75% concordance with mNGS for pathogen detection; high diagnostic accuracy for bloodstream infections [87] |
Representative Experimental Protocol: Ultra-Broad Hybrid Capture for Pathogen Detection
This protocol, adapted from a 2025 study on bloodstream infections, demonstrates how targeted sequencing can be scaled for broad detection panels [87]:
- Cell-free DNA (cfDNA) Extraction: Extract cfDNA from patient plasma using a commercial kit.
- Library Preparation: Construct a sequencing library from the extracted cfDNA using end-repair, adapter ligation, and amplification.
- Hybrid Capture Enrichment: Incubate the pre-library with an ultra-broad pathogen panel (e.g., covering 1,872 microbial species) using high-density tiling probes for 4 hours. This step enriches pathogen-derived sequences.
- Washing and Elution: Remove non-specifically bound fragments and elute the enriched library.
- Sequencing: Sequence on a platform like the Gene+ Seq-100 with a relatively low depth of 5 million reads.
- Bioinformatic Analysis: Process data through an automated pipeline. Normalize reads to reads per million (RPM) and apply reporting thresholds (e.g., RPM ≥6 for common bacteria) to identify causative pathogens [87].
This targeted approach demonstrated diagnostic accuracy comparable to metagenomic NGS (mNGS) but at a significantly lower cost and sequencing depth [87].
Successful implementation of hybrid and targeted approaches relies on key reagents, technologies, and software tools.
Table 3: Essential Resources for Hybrid and Targeted Sequencing
| Category | Item | Function & Application |
|---|---|---|
| Commercial Panels | 10x Genomics Chromium Single Cell 3' Kit [4] | Enables single-cell RNA-seq library generation from partitioned cells, compatible with both short- and long-read sequencing of the same cDNA. |
| MAS-ISO-seq for 10x Genomics (PacBio) [4] | A targeted long-read approach that concatenates transcripts for efficient sequencing on PacBio platforms, allowing for isoform-resolution in single cells. | |
| Ultra-broad Hybrid Capture Panels (e.g., 1872 pathogens) [87] | Designed for sensitive detection of a wide range of pathogens from cfDNA, making them suitable for diagnosing complex infections like bloodstream infections. | |
| Bioinformatics Tools | nf-core/nanoseq [6] | A community-curated, standardized pipeline for processing long-read RNA-seq data, including QC, alignment, transcript quantification, and differential expression. |
| SQANTI3 [4] | A tool for quality control and classification of long-read transcripts against a reference annotation. | |
| BLAZE, Scywalker [4] | Software tools for processing barcoded long-read single-cell data. | |
| Reference Materials | Genome in a Bottle (GIAB) Benchmarks [86] | Authoritative reference genomes with highly characterized variant calls, essential for validating the performance of sequencing pipelines in challenging genomic regions. |
| SPIKE-IN RNAs (e.g., SIRVs, Sequins) [6] | RNA molecules with known sequences and concentrations spiked into samples to quantitatively evaluate the accuracy, sensitivity, and dynamic range of transcriptomic assays. |
The choice between short-read, long-read, hybrid, and targeted sequencing is not a matter of selecting a single superior technology. Instead, it is a strategic decision based on the specific biological question, required resolution, and available resources. Short-read sequencing remains the workhorse for high-throughput, quantitative gene expression studies. Long-read sequencing is transformative for resolving isoform complexity, discovering novel transcripts, and interrogating challenging genomic regions. Hybrid approaches offer the most comprehensive view by integrating the strengths of both. Targeted methods provide a cost-effective and highly sensitive solution for focused applications, especially in clinical diagnostics.
As these technologies continue to evolve, becoming more accurate and affordable, their integrated use will undoubtedly deepen our understanding of transcriptomic and genomic complexity, accelerating discovery in basic research and drug development.
Head-to-Head Performance: Validating Accuracy in Genomic and Clinical Contexts
The accurate identification of genetic variants—including single nucleotide variants (SNVs), small insertions and deletions (indels), and structural variants (SVs)—is a cornerstone of modern genomics research, with critical applications in drug development and disease mechanism elucidation. For years, short-read sequencing (e.g., Illumina) has been the dominant technology, providing high-throughput, cost-effective data that has enabled massive genomics projects. However, a paradigm shift is underway with the rise of long-read sequencing technologies from PacBio and Oxford Nanopore Technologies (ONT). These technologies can sequence DNA or RNA molecules thousands to tens of thousands of bases long, overcoming the inherent limitation of short reads in resolving repetitive regions and mapping unique splice junctions [10]. This guide objectively compares the performance of variant calling tools across these platforms, framing the discussion within the broader thesis of short-read versus long-read sequencing for comprehensive genomic variant discovery.
Experimental Protocols for Benchmarking Variant Callers
Benchmarking studies rely on robust experimental designs and well-characterized samples to establish "ground truth." The following methodologies are commonly employed in the field.
Use of Established Reference Materials and Spike-Ins
A critical strategy involves sequencing biological reference samples with known or partially known variant profiles.
- Genome in a Bottle (GIAB) Consortium: Provides a highly characterized benchmark genome (HG002) for both small variants and SVs. This is used to assess the precision and recall of calling tools on real data [89].
- Spike-in Control RNAs: Synthetic RNA sequences with known concentrations and structures, such as those from the External RNA Control Consortium (ERCC) or the SIRV and Sequin sets, are added to samples before library preparation [6] [90] [91]. This creates an internal, absolute standard for evaluating the accuracy of expression quantification and isoform detection in RNA-seq data.
- Cell Line Mixtures: Creating in silico or laboratory mixtures of RNA from two different cell lines (e.g., the MAQC samples or the Quartet project samples) provides known expression fold-changes against which the accuracy of differential expression analysis can be measured [90] [91].
The Tumor-Normal Somatic Variant Calling Workflow
For somatic variant discovery in cancer, a common and rigorous protocol involves paired tumor and normal samples. A representative workflow for identifying somatic structural variants is detailed below [92]:
- Sample Preparation & Sequencing: DNA is extracted from both a tumor sample and a matched normal sample from the same patient. Both are sequenced using the same long-read platform (e.g., PacBio or ONT).
- Quality Control & Alignment: Raw sequencing data is assessed for quality (using tools like FASTQC). Reads are then aligned to a reference genome (e.g., GRCh38) using a long-read aware aligner such as
minimap2. - Variant Calling: Structural variant callers (e.g.,
Sniffles2,cuteSV,Delly) are run separately on the tumor and normal BAM files to generate initial variant call format (VCF) files. - Somatic SV Identification: Two primary methods are used:
- Subtraction Method: The normal sample's VCF is used as a filter to remove germline variants, leaving candidate somatic SVs present only in the tumor.
- Direct Somatic Calling: Specialized tools like
Severusare designed to analyze the tumor and normal data together to directly output somatic SV calls.
- Validation: Candidate somatic SVs are often manually curated and validated using tools like the Integrative Genomics Viewer (IGV) or against an established truth set, such as the one available for the COLO829 melanoma cell line [92].
The diagram below visualizes the standard workflow for identifying somatic structural variants from long-read sequencing data of paired tumor-normal samples.
Performance Comparison of Variant Calling Tools
Structural Variant Calling
Structural variants (SVs), defined as genomic alterations ≥50 base pairs, are major drivers of disease but have been notoriously difficult to detect with short reads. Long-read sequencing has significantly improved this area. The table below summarizes benchmark findings for SV callers across sequencing technologies.
Table 1: Benchmarking Performance of Structural Variant Callers
| Sequencing Tech. | Top-Performing Tool(s) | Key Performance Findings | Study/Context |
|---|---|---|---|
| Short-Read (Illumina) | DRAGEN v4.2 | Highest accuracy among 10 srWGS callers tested [89]. | HG002 Benchmark Deletions [89] |
| Manta (with minimap2) | Achieved performance comparable to DRAGEN [89]. | HG002 Benchmark Deletions [89] | |
| PacBio Long-Read | Sniffles2 | Outperformed other tested tools [89]. | HG002 Benchmark Deletions [89] |
| ONT Long-Read | Dysgu (high coverage)Duet (≤10x coverage) | Best results at high coverage.Highest accuracy at low coverage [89]. | HG002 Benchmark Deletions [89] |
| Multiple Long-Read | Combination of multiple callers | Combining tools like Sniffles, cuteSV, etc., significantly enhances true somatic SV detection accuracy [92]. | Somatic SV detection in cancer [92] |
A critical finding from recent benchmarks is that the choice of alignment software significantly impacts SV calling from both short and long-read data, an factor sometimes overlooked. For short-read data, using minimap2 with Manta achieved performance comparable to the commercial DRAGEN pipeline. For ONT data, minimap2 consistently led to the best results among the aligners tested [89].
Transcript-Level Analysis and Isoform Detection
In RNA sequencing, a primary goal is to accurately identify and quantify the full-length isoforms expressed from each gene. This has been a fundamental challenge for short-read data. The table below compares the performance of tools for transcript discovery and differential expression.
Table 2: Benchmarking Performance of Long-Read RNA-Seq Analysis Tools
| Analysis Task | Top-Performing Tool(s) | Key Performance Findings | Study/Context |
|---|---|---|---|
| Isoform Detection | StringTie2, Bambu | Outperformed four other tested tools for identifying full-length transcripts [91]. | In silico mixtures with spike-in sequins [91] |
| Differential Transcript Expression (DTE) | DESeq2, edgeR, limma-voom | These established short-read tools performed best for DTE analysis even on long-read data [91]. | In silico mixtures with spike-in sequins [91] |
| Differential Transcript Usage (DTU) | No clear front-runner | Further methods development is needed for this application [91]. | In silico mixtures with spike-in sequins [91] |
| Gene Expression Quantification | HTSeq, Cufflinks, RSEM, IsoEM | HTSeq showed highest correlation with RT-qPCR (0.89), but others may have higher absolute accuracy [93]. | MAQC samples with RT-qPCR validation [93] |
A key insight from the Singapore Nanopore Expression (SG-NEx) project is that different long-read RNA-seq protocols (Direct RNA, Direct cDNA, PCR-cDNA) introduce specific biases. PCR-amplified cDNA sequencing generates the highest throughput but can over-represent highly expressed genes. In contrast, PCR-free protocols preserve a broader transcript diversity but with lower yield [6].
The Impact of Experimental and Bioinformatics Factors
Large-scale, real-world benchmarking studies reveal that technical variation is a major challenge. A multi-center study using the Quartet and MAQC reference materials found significant inter-laboratory variation in RNA-seq results, especially when trying to detect subtle differential expression—a common scenario in clinical diagnostics comparing different disease subtypes or stages [90].
The primary sources of this variation were traced to specific steps in the workflow:
- Experimental Factors: mRNA enrichment method and library strandedness were major contributors to variation [90].
- Bioinformatics Factors: Every step in the computational pipeline, from the choice of gene annotation file to the alignment tool, quantification method, and normalization strategy, introduced significant variability. The study concluded that no single tool is superior in all scenarios, emphasizing the need for careful pipeline selection based on the specific biological question [90].
To ensure reproducible and accurate variant calling, researchers rely on a suite of well-characterized reagents and data resources.
Table 3: Essential Resources for Benchmarking Variant Calling
| Resource | Type | Primary Function in Benchmarking |
|---|---|---|
| GIAB HG002 | Reference Genome | Provides a benchmark set of validated variants for assessing caller accuracy and recall [89]. |
| ERCC Spike-Ins | Synthetic RNA Controls | Act as an internal standard with known concentration to evaluate quantification accuracy [90]. |
| SIRV/Sequin Spike-Ins | Synthetic RNA Controls | Isoform-level spike-in controls with complex sequences for validating transcript identification and quantification [6] [91]. |
| MAQC & Quartet Samples | Biological Reference Materials | Cell line RNAs with well-studied expression profiles for cross-platform and cross-laboratory reproducibility studies [90]. |
| NF-Core Pipelines (e.g., Nanoseq) | Bioinformatics Workflow | Community-curated, standardized pipelines for processing long-read RNA-seq data to ensure consistent and reproducible analyses [6]. |
The comprehensive benchmarking of variant calling tools underscores a clear trend: while short-read technologies and their associated callers remain highly accurate and cost-effective for SNV and small indel detection, long-read technologies are indispensable for the comprehensive discovery of structural variants and the full-length characterization of RNA isoforms. The choice between them is no longer binary; many sophisticated research and diagnostic pipelines now leverage the strengths of both in a complementary manner.
For structural variants, the combination of long-read sequencing with a multi-caller approach (e.g., using Sniffles2, cuteSV, and Delly in parallel) provides the most robust detection, especially for somatic variants in cancer [92] [89]. For transcriptomics, long-read sequencing directly resolves isoforms, and while specialized tools like Bambu and StringTie2 excel at discovery, established differential expression tools like DESeq2 and edgeR remain powerful for quantification even on long-read data [6] [91]. As the field moves toward clinical application, the community-driven development of standardized resources—from reference materials like the Quartet sets to computational pipelines like nf-core/nanoseq—will be critical for ensuring the accuracy and reproducibility required for drug development and future clinical diagnostics.
The accurate characterization of medically relevant genes is fundamental to advancing genomic research and precision medicine. However, a significant portion of the human genome, including complex repetitive regions and genes with highly homologous pseudogenes, has historically challenged conventional short-read sequencing technologies, leading to potential gaps in diagnostic data [94]. The emergence of long-read sequencing platforms from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) promises to overcome these limitations by providing the read length necessary to span repetitive elements and resolve complex structural variations [95] [10]. This guide provides an objective, data-driven comparison of short-read and long-read sequencing performance for analyzing challenging genomic regions critical to human health.
Key Technological Comparisons
The core difference between short-read and long-read technologies lies in read length and library preparation. Short-read platforms (e.g., Illumina) generate fragmented data (75-300 bp reads) requiring complex computational assembly, which falters in repetitive zones [10]. In contrast, long-read technologies produce reads spanning kilobases to megabases, enabling direct sequencing through repetitive elements and complex structural variants [95] [10].
Table 1: Core Technology Comparison of Leading Sequencing Platforms
| Feature | Short-Read (Illumina) | Long-Read (PacBio HiFi) | Long-Read (ONT) |
|---|---|---|---|
| Typical Read Length | 75-300 bp | 10-25 kb | 20 kb -> 1 Mb+ |
| Single-Base Accuracy | >99.9% (Q30+) | >99.9% (Q30-Q40) [95] [10] | ~98-99.5% (Q20+ chemistry) [95] |
| Primary Strengths | High throughput, low per-base cost, established workflows | High accuracy, excellent for SV detection and phasing [95] | Ultra-long reads, real-time analysis, portability [95] |
| Major Challenge | Limited resolution in repeats and SVs [94] | Higher cost per genome, shorter reads than ONT [95] | Historically lower accuracy (improving with new chemistry) [95] |
PacBio's High Fidelity (HiFi) sequencing uses circular consensus sequencing (CCS) to achieve >99.9% accuracy by repeatedly reading the same DNA molecule [95]. ONT technology identifies nucleotides as single DNA molecules pass through a protein nanopore, enabling ultra-long reads but with a slightly higher native error rate that is mitigated by new chemistries and basecalling algorithms [95].
Performance in Challenging Medically Relevant Regions
Comparative studies demonstrate that long-read technologies significantly outperform short-read approaches in resolving structurally complex genomic regions. A landmark 2025 study sequenced 65 diverse human genomes to telomere-to-telomere (T2T) status, closing 92% of previous assembly gaps and completely resolving 1,852 complex structural variants and 1,246 human centromeres that were previously intractable [96] [97]. This research highlights complete sequence continuity at multiple complex loci:
- SMN1/SMN2: Critical for spinal muscular atrophy diagnosis and therapy, these genes are highly homologous and difficult to resolve with short reads [96] [97].
- Major Histocompatibility Complex (MHC): Associated with cancer, autoimmune diseases, and over 100 other conditions [97].
- NBPF8: Involved in developmental and neurogenetic disease [97].
- AMY1/AMY2: The amylase gene cluster important for starch digestion [97].
Table 2: Performance Comparison for Specific Challenging Loci
| Genomic Region / Challenge | Clinical Relevance | Short-Read Performance | Long-Read Performance |
|---|---|---|---|
| Pseudogenes (e.g., NCF1) | Chronic granulomatous disease [98] | Mis-mapping to homologous pseudogenes causes false positives/negatives [94] | Accurately distinguishes functional genes from pseudogenes [98] |
| Repeat Expansions (e.g., DMPK) | Myotonic dystrophy [98] | Limited ability to size large repeats | Fully resolves large repeat expansions [98] |
| Centromeric Regions | Cell division, essential structures | Highly fragmented or absent in assemblies [96] | Completely assembled and validated [96] |
| Mobile Element Insertions | Genomic instability, disease | Limited detection | Identified 12,919 MEIs across 130 haplotypes [96] |
Experimental Data and Benchmarking
Structural Variant Detection in Rare Diseases
Long-read sequencing has demonstrated transformative potential for diagnosing rare genetic diseases, which are often caused by structural variants (SVs) difficult to detect with short-read technologies. Benchmarking studies show that PacBio HiFi consistently achieves F1 scores greater than 95% for SV detection, while ONT excels at resolving larger, more complex rearrangements [95]. Following inconclusive short-read sequencing, PacBio HiFi whole-genome sequencing increased diagnostic yield by 10-15% in rare disease populations, uncovering cryptic SVs, phasing-dependent compound heterozygous mutations, and repetitive expansions [95].
Targeted Sequencing in Cancer Genomics
A 2025 methodological comparison on colorectal cancer (CRC) samples provided direct performance metrics across platforms [7]. The study evaluated key cancer genes including KRAS, BRAF, TP53, APC, and PIK3CA using both Illumina whole-exome and Nanopore whole-genome sequencing.
Table 3: Quantitative Performance Metrics from Colorectal Cancer Study [7]
| Performance Metric | Illumina Short-Read | Nanopore Long-Read |
|---|---|---|
| Average Coverage Depth | 105.88X ± 30.34X | 21.20X ± 6.60X (CRC samples) |
| Median Mapping Quality (Phred) | 33.67 (99.96% accuracy) | 29.8 (99.89% accuracy) |
| SV Analysis | Limited resolution of complex rearrangements | Enhanced ability to resolve large/complex SVs with high precision |
While Illumina showed slightly higher mapping quality and coverage depth in this study, Nanopore sequencing provided superior resolution of structural variants and complex genomic rearrangements relevant to cancer pathogenesis [7].
RNA Sequencing and Isoform Resolution
For transcriptome analysis, long-read RNA sequencing enables full-length transcript characterization without assembly, directly revealing alternative splice variants, fusion transcripts, and allele-specific expression. The Singapore Nanopore Expression (SG-NEx) project conducted a comprehensive benchmark comparing five RNA-seq protocols across seven human cell lines [6]. The study found that PCR-amplified cDNA sequencing (Nanopore) and PacBio IsoSeq showed the most uniform coverage across transcript length and the highest proportion of reads spanning all exon junctions ("full-splice-match reads") [6]. Long-read protocols specifically provided more robust identification of major isoforms and complex transcriptional events [6].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Research Reagents and Solutions for Sequencing Complex Regions
| Reagent/Solution | Function/Application | Example Use Cases |
|---|---|---|
| PacBio HiFi Sequencing Kits | Generate highly accurate long reads (10-25 kb, >99.9% accuracy) | SV detection in rare disease [95], haplotype phasing [96] |
| ONT Ultra-Long DNA Kits | Produce reads >100 kb for spanning massive repeats | Assembling centromeres [96], resolving complex SVs [95] |
| Illumina Constellation Kits | Emerging technology to access difficult regions on short-read platforms | Detecting variants in SMN1, NCF1, and DMPK [98] |
| Strand-Seq | Provides phasing information for assembly | Global phasing of assembly graphs [96] |
| Bionano Optical Mapping | Generates long-range genome maps for validation | Scaffolding and validating assemblies [96] |
| Spike-in RNA Controls (e.g., SIRV, ERCC) | Assess technical performance and quantification accuracy | Benchmarking RNA-seq protocol performance [6] |
Experimental Workflows and Methodologies
Comprehensive Genome Assembly Workflow
The recent Human Genome Structural Variation Consortium study that produced 130 haplotype-resolved assemblies exemplifies a robust methodology for resolving complex regions [96]. The workflow integrated multiple complementary technologies:
Workflow for Comprehensive Genome Assembly. This multi-platform approach combines accurate PacBio HiFi reads, ultra-long ONT reads, and phasing data from Strand-seq to generate complete, haplotype-resolved assemblies. T2T: telomere-to-telomere; SV: structural variant.
This methodology achieved remarkable results: 602 chromosomes were assembled as single gapless contigs from telomere to telomere, with 92% of previous assembly gaps closed [96]. The assemblies enabled identification of 26,115 structural variants per individual - a substantial increase amenable to downstream disease association studies [96].
Resolving the Pseudogene Challenge
The challenge of pseudogenes arises when highly homologous sequences cause mis-mapping of sequencing reads. Blueprint Genetics outlines how homology levels affect analysis [94]:
Pseudogene Analysis Challenge. This diagram illustrates how long-read sequencing overcomes mis-mapping issues in highly homologous regions by providing unique flanking sequences for alignment.
When homology exceeds 98%, specialized methods become essential. Illumina's emerging Constellation technology aims to address this within short-read frameworks by using proximity-based mapping to resolve ambiguities in regions like SMN1/SMN2 and NCF1 [98].
The evidence demonstrates that long-read sequencing technologies provide transformative capabilities for analyzing medically relevant genes in complex and repetitive regions. While short-read sequencing remains the workhorse for many applications due to its cost-effectiveness and high base-level accuracy, it exhibits fundamental limitations in resolving structural variants, pseudogenes, and repetitive elements. PacBio HiFi and ONT platforms now enable complete characterization of previously intractable regions like centromeres, segmental duplications, and complex structural variants, with demonstrated diagnostic utility in rare diseases and cancer. As these technologies continue to evolve with improving accuracy and declining costs, they are poised to become indispensable tools for comprehensive genomic analysis in both research and clinical settings.
The fundamental choice between short-read and long-read sequencing technologies is pivotal in designing modern transcriptomics studies. For years, short-read RNA sequencing has been the established backbone for gene expression profiling, offering high throughput and cost-effectiveness for quantifying gene-level expression [4] [9]. However, its limitations in resolving complex isoforms have persisted as a significant challenge. The emergence of sophisticated long-read technologies from PacBio and Oxford Nanopore Technologies (ONT) now enables full-length transcript sequencing, revealing a previously inaccessible layer of transcriptomic complexity involving alternative splicing, novel isoforms, and sequence variations [4] [8]. This guide provides a direct, data-driven comparison of these platforms, framing their performance within the broader thesis of how researchers can strategically select technologies based on specific biological questions. We synthesize evidence from recent, rigorous benchmarking studies to objectively evaluate platforms based on accuracy, throughput, and applicability, providing a framework for researchers, scientists, and drug development professionals to navigate this evolving technological landscape.
Technology Platform Comparison at a Glance
The table below summarizes the core technical specifications and performance characteristics of major short-read and long-read RNA sequencing platforms, based on recent comparative studies and benchmarking data.
Table 1: Direct Comparison of RNA Sequencing Technologies
| Feature | Illumina (Short-Read) | PacBio (Long-Read) | Oxford Nanopore (Long-Read) |
|---|---|---|---|
| Sequencing Principle | Sequencing by Synthesis (SBS) [9] | HiFi Read via Circular Consensus Sequencing [9] | Nanopore current modulation [9] |
| Typical Read Length | 50-300 bp [9] | Full-length transcripts (>10 kb common) [6] [99] | Full-length transcripts [6] |
| Throughput | Very high (cost-effective per base) [9] | High (improved with Kinnex) [16] [99] | High (PCR-cDNA protocol) [6] |
| Key Strengths | High gene-level quantification accuracy, mature analytics, low cost per sample [4] [9] | High single-molecule accuracy, excellent isoform resolution, low inference variability [16] [99] | Direct RNA sequencing, detection of RNA modifications, real-time analysis [6] [9] |
| Primary Limitations | Cannot resolve complex isoforms; inference challenges for transcript-level quantification [6] [99] | Historically lower throughput; higher input requirements for some applications | Higher raw read error rate requiring computational correction [6] |
| Ideal Use Cases | Bulk gene expression studies, large cohort screening, SNP/small variant detection [9] | Differential transcript expression, novel isoform discovery, allele-specific expression, genome annotation [16] [99] [100] | RNA modification detection (e.g., m6A), rapid diagnostic applications, direct RNA sequencing [6] |
Experimental Data and Performance Benchmarking
Recent independent benchmarks and large-scale consortium studies provide rigorous performance data comparing these platforms.
Quantification Accuracy and Technical Reproducibility
A cornerstone of reliable transcriptomics is the ability of a platform to accurately quantify expression and yield reproducible results across technical replicates. Evidence from matched-sample comparisons reveals distinct performance profiles.
- Gene-Level Concordance: At the gene level, both short-read and long-read technologies show strong concordance. A study sequencing the same 10x Genomics 3' cDNA libraries with both Illumina and PacBio platforms found that "both methods render highly comparable results" for gene-level counts [4]. Similarly, the SG-NEx project reported that "Gene expression is robustly estimated across protocols" when analyzing spike-in RNA controls with known concentrations [6].
- Transcript-Level Divergence: The critical advantage of long-read sequencing emerges at the transcript level. PacBio Kinnex data demonstrated "Pearson correlations exceeding 0.9 at the gene level and approaching 0.9 at the transcript level" when compared to Illumina, indicating high concordance for resolvable transcripts [16]. However, Illumina data exhibited "substantially higher inferential variability" (replicate-to-replicate fluctuations) and "transcript flips" for complex genes, where quantification of nearly identical isoforms was inconsistent across replicates [16] [99]. In contrast, Kinnex provided stable and consistent quantification, leading to more reliable detection of differential transcript expression (DTE) [99].
- Spike-In Analysis: The SG-NEx project's use of spike-in RNAs (ERCC, SIRV) with known concentrations provided a ground truth for assessing quantification accuracy. Their analysis found that "Nanopore long-read RNA-seq data showed the lowest estimation error overall and a higher correlation with the expected concentrations" compared to other protocols, including short-read and PacBio Iso-Seq [6].
Discovery Power and Resolution of Complex Loci
The ability to discover novel biological features is a key differentiator. Long-read sequencing uniquely enables the characterization of complex transcriptional events that are intractable for short-read methods.
- Novel Isoform and Splice Site Discovery: Applications in non-model organisms and complex tissues consistently reveal extensive unannotated transcriptomic diversity. A study in Atlantic salmon using ONT long-read RNA-seq generated a transcriptome where 60% of transcript models contained a novel splice site, a five-fold increase in the transcript-to-gene ratio compared to the existing Ensembl reference [100]. In human oocytes, PacBio Iso-Seq revealed that "about 40%" of isoforms were novel transcripts not found in the GENCODE reference, a finding "underestimated" by short-read sequencing [16].
- Resolution of Complex Genes: Short reads struggle to span multiple splice junctions in a single fragment, leading to ambiguous mapping. This results in an artificial "division" of expression among similar isoforms, making it difficult to determine the true expressed transcript [99]. Long reads, by capturing the entire transcript in a single read, eliminate this ambiguity, providing a direct and accurate picture of isoform expression in genes with complex architecture.
- Detection of Other Transcriptomic Features: Long-read technologies excel at identifying structural variations, fusion transcripts, and repetitive elements. ONT's direct RNA sequencing can detect RNA modifications like N6-methyladenosine (m6A) [6], while PacBio's HiFi reads have been used to identify and phase allele-specific splicing events [16]. The SG-NEx project also highlighted long-read sequencing's superior ability to identify full-length fusion transcripts that short-read methods often miss [101].
Detailed Experimental Protocols for Technology Benchmarking
To ensure the validity and reproducibility of the comparative data discussed, the cited studies implemented rigorous and detailed experimental methodologies.
Cross-Platform Comparison from a Shared cDNA Library
A key methodology for direct, bias-free comparison involves sequencing the same cDNA library on different platforms.
- Library Source: In one benchmark, researchers used the same full-length cDNA generated from the 10x Genomics Chromium Single Cell 3' Reagent Kit (v3.1 Chemistry) for both Illumina and PacBio sequencing [4].
- Illumina Library Prep: The shared cDNA was enzymatically sheared to 200-300 bp, and standard Illumina sequencing libraries were constructed with end repair, A-tailing, adapter ligation, and sample index PCR. Sequencing was performed on an Illumina NovaSeq 6000 [4].
- PacBio (MAS-ISO-seq/Kinnex) Library Prep: The same cDNA (45 ng input) was used for PacBio's MAS-ISO-seq (now Kinnex) library preparation. This involved a specific PCR step with a modified primer to remove template-switching oligonucleotide (TSO) artefacts, followed by concatenation of cDNA molecules into longer fragments (10-15 kb) for efficient sequencing on the PacBio Sequel IIe system [4].
- Analysis for Cross-Comparison: Crucially, each cDNA molecule was tagged with a unique cell barcode and unique molecular identifier (UMI), allowing for a per-molecule comparison between the platforms by matching these tags [4].
The SG-NEx Multi-Protocol Benchmarking Framework
The Singapore Nanopore Expression (SG-NEx) project established one of the world's most comprehensive benchmarking resources.
- Cell Lines and Replicates: The core dataset consists of seven human cell lines (e.g., HCT116, HepG2, A549), each sequenced with a minimum of three high-quality replicates across multiple platforms [6] [101].
- Sequencing Protocols: Each cell line was profiled using:
- Illumina short-read RNA-seq (paired-end, 150 bp).
- Nanopore direct RNA-seq (dRNA).
- Nanopore amplification-free direct cDNA (d cDNA).
- Nanopore PCR-amplified cDNA (cDNA).
- PacBio Iso-Seq (for a subset) [6].
- Spike-Ins and Controls: The study incorporated multiple spike-in RNA standards (Sequin, ERCC, SIRVs) with known concentrations, enabling absolute quantification and accuracy assessment [6].
- Standardized Bioinformatics: The project developed and utilized the nf-core/nanoseq community-curated pipeline to ensure consistent and reproducible data processing across all samples and technologies. This pipeline handles quality control, alignment, transcript quantification, and differential expression analysis [6].
Visualizing the Sequencing and Analysis Workflow
The following diagram illustrates the core experimental and computational steps for a cross-platform benchmarking study, as implemented in the methodologies described above.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful execution of a comparative transcriptomics study relies on a suite of specialized reagents and computational tools. The table below details essential components used in the featured experiments.
Table 2: Essential Reagents and Tools for RNA-Seq Benchmarking
| Item | Function | Example Use-Case |
|---|---|---|
| 10x Genomics 3' Reagent Kits | Generates barcoded full-length cDNA from single cells or bulk RNA, enabling parallel sequencing on different platforms. | Creating a shared cDNA library for direct Illumina/PacBio comparison [4]. |
| PacBio MAS-ISO-Seq/Kinnex Kit | Prepares cDNA for long-read sequencing by removing artifacts and concatenating transcripts for high throughput. | Enabling high-depth long-read transcriptome profiling for quantification [4] [16]. |
| Spike-in RNA Controls (ERCC, SIRV, Sequin) | Provides an internal standard with known concentration for assessing quantification accuracy and technical variability. | Benchmarking platform performance and normalization accuracy in the SG-NEx project [6]. |
| Ribosomal Depletion Kits | Removes abundant ribosomal RNA (rRNA) to increase the proportion of informative reads in total RNA sequencing. | Enhancing coverage of mRNA and non-polyadenylated RNAs; critical for degraded samples [32]. |
| Stranded Library Prep Kits | Preserves the original orientation of transcripts during cDNA synthesis, crucial for identifying antisense transcription and accurately quantifying overlapping genes. | Essential for novel lncRNA discovery and correct interpretation of splicing patterns [32]. |
| nf-core/nanoseq Pipeline | A community-curated, standardized bioinformatics workflow for processing long-read and short-read RNA-seq data. | Ensuring reproducible and comparable analysis across different technologies and studies [6]. |
The evidence from recent, rigorous benchmarks indicates that the choice between short-read and long-read RNA sequencing is no longer a simple question of which technology is superior, but rather which is fit-for-purpose for a specific biological question. Short-read sequencing (Illumina) remains a powerful and cost-effective tool for projects focused on gene-level differential expression in large cohorts or the detection of small genetic variants. Its maturity, high throughput, and low cost per sample make it ideal for initial screening and bulk expression analysis.
In contrast, long-read sequencing (PacBio and ONT) has matured into an indispensable technology for research that demands isoform-level resolution. PacBio Kinnex, with its high accuracy and low inferential variability, is a reliable choice for differential transcript expression analysis, novel isoform discovery, and allele-specific expression studies [16] [99]. ONT sequencing offers unique capabilities in direct RNA sequencing and the detection of RNA modifications, providing insights into the epitranscriptome [6]. The decision-making framework for platform selection should therefore be guided by the research objective: opt for short-reads for high-throughput gene counting, and invest in long-reads to unravel the full complexity of the transcriptome, especially in the context of disease research, genome annotation, and developmental biology.
Large-scale genomic initiatives are powerful engines for biological discovery, and the choice of sequencing technology is fundamental to the insights they can generate. Using the All of Us Research Program as a primary case study, this guide examines how the strategic application of short-read and long-read RNA sequencing technologies shapes research outcomes, providing objective performance data to inform your own experimental plans.
The All of Us Research Program: A Paradigm of Scale and Diversity
The All of Us Research Program is a landmark longitudinal cohort study in the United States aiming to enroll at least one million participants to accelerate biomedical research and improve human health [102]. Its design directly addresses a critical historical limitation in the field: the severe under-representation of large subsets of individuals in biomedical research [102].
A key to its success is the generation of clinical-grade whole-genome sequence (WGS) and genotyping data. In its 2024 data release, the program included 245,388 clinical-grade genome sequences [102]. The resource is unparalleled in its diversity:
- 77% of participants are from communities that are historically under-represented in biomedical research.
- 46% are from under-represented racial and ethnic minorities [102].
By linking this diverse genomic data to longitudinal electronic health records (EHRs), available for over 287,000 participants, the program creates a rich dataset for validating genetic associations across ancestries. This has enabled the replication of associations for 3,724 genetic variants linked to 117 diseases in both participants of European and African ancestry with high replication rates [102]. The program's data is accessible to researchers through the Researcher Workbench, with a median time from registration to data access of just 29 hours [102].
Sequencing Technologies: A Technical and Performance Comparison
The choice between short-read and long-read sequencing involves trade-offs. The table below summarizes the core characteristics of each approach.
Table 1: Core Characteristics of RNA Sequencing Technologies
| Feature | Short-Read RNA-Seq | Long-Read RNA-Seq |
|---|---|---|
| Representative Platforms | Illumina, Ion Torrent [30] | PacBio, Oxford Nanopore [30] |
| Typical Read Length | 50-600 base pairs [10] | 5,000 - 30,000+ base pairs [10] |
| Primary Strengths | High accuracy, cost-effectiveness, scalability, high throughput; ideal for gene-level expression and SNP detection [30]. | Resolves complex genomic structures, identifies full-length transcript isoforms, detects structural variations, and can reveal RNA modifications [30] [79]. |
| Key Limitations | Limited ability to resolve repetitive regions, phase haplotypes, or quantify specific alternative transcript isoforms [79]. | Historically higher cost and error rates, though accuracy has dramatically improved (e.g., PacBio HiFi reads at >99.9% accuracy) [10]. |
| Ideal Applications | Differential gene expression (DGE) analysis, small RNA sequencing, single-cell analysis, SNP detection [30]. | Isoform discovery, ab initio transcriptome analysis, fusion transcript detection, complex transcript analysis (e.g., MHC, HLA) [30]. |
Recent systematic benchmarks, such as the Singapore Nanopore Expression (SG-NEx) project, provide rigorous, data-driven performance comparisons. This study profiled seven human cell lines using five different RNA-seq protocols, including short-read cDNA sequencing and multiple Nanopore long-read protocols (direct RNA, direct cDNA, and PCR-cDNA) [6].
Table 2: Experimental Findings from the SG-NEx Benchmarking Study [6]
| Performance Metric | Key Findings |
|---|---|
| Throughput & Read Length | PCR-amplified cDNA long-read sequencing achieved throughput matching short-read RNA-seq. PacBio IsoSeq generated the longest reads on average. |
| Transcript Coverage | Long-read protocols showed more uniform coverage across the 5' and 3' ends of transcripts compared to short-read data. PCR-amplified cDNA and PacBio data had the highest proportion of reads spanning all exon junctions. |
| Quantification Accuracy | For overall gene-level expression, both short-read and long-read data showed strong correlation with known spike-in RNA concentrations. |
| Protocol Bias | PCR-amplified cDNA protocols showed a bias toward highly expressed genes, while PacBio data showed a significant depletion of shorter transcripts. |
Experimental Protocols in Practice
All of Us Whole-Genome Sequencing Protocol
The All of Us program employs a centralized, standardized pipeline to ensure data quality and clinical-grade precision [102].
- Sample Preparation: Blood-derived DNA from participants is used to create PCR-free barcoded WGS libraries with the Illumina Kapa HyperPrep kit.
- Sequencing: Libraries are pooled and sequenced on the Illumina NovaSeq 6000 instrument.
- Quality Control & Processing: Initial QC is performed with the Illumina DRAGEN pipeline, assessing contamination, mapping quality, and concordance with genotyping array data. The Data and Research Center performs further QC and joint calling across the entire dataset.
- Variant Discovery & Annotation: The program developed a cloud-based Genomic Variant Store (GVS) to manage the massive scale of data, identifying over 1 billion genetic variants, including 275 million previously unreported variants [102]. Variants are annotated using Illumina Nirvana.
SG-NEx Multi-Protocol Long-Read RNA Sequencing
The SG-NEx project provides a community-curated pipeline (nf-core/nanoseq) for streamlined long-read data analysis [6]. Its experimental workflow for comparing protocols is outlined below.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Genomic Sequencing
| Item | Function in the Protocol |
|---|---|
| Illumina Kapa HyperPrep Kit | Used in All of Us for constructing PCR-free, barcoded WGS libraries to minimize amplification bias [102]. |
| PacBio Iso-Seq Express 2.0 Kit | Used for synthesizing and amplifying high-quality cDNA from total RNA in long-read isoform sequencing studies [37]. |
| Oxford Nanopore Direct RNA Sequencing Kit | Enables sequencing of native RNA, bypassing cDNA synthesis to allow direct detection of RNA modifications [6]. |
| Spike-in RNA Controls (e.g., ERCC, SIRV, Sequin) | Artificial RNA sequences with known concentrations spiked into samples to evaluate quantification accuracy and technical variability across protocols [6]. |
| PAXgene Blood RNA Tubes | Specialized collection tubes for stabilizing RNA in whole blood samples at the point of collection, crucial for clinical and biobank studies [37]. |
| Agilent RNA 6000 Nano Kit | Used with the Bioanalyzer instrument to assess RNA Integrity Number (RIN), a critical quality control step before library preparation [103]. |
Large-scale initiatives like All of Us demonstrate the immense power of high-quality, diverse genomic data coupled with deep phenotypic information. The strategic selection of sequencing technology is paramount:
- For projects requiring massive scale, high accuracy, and cost-effective genotyping or gene-level expression analysis, short-read sequencing remains the workhorse technology.
- When the research goal is to unravel transcriptional complexity, discover novel isoforms, detect gene fusions, or study RNA modifications, long-read RNA sequencing is indispensable.
The evolving landscape, with platforms like PacBio's Revio and Illumina's Complete Long-Reads, is making long-read technology more accessible. The future lies in hybrid approaches, leveraging the high sensitivity of short-read data with the comprehensive resolution of long-read data to gain a complete picture of the genome and transcriptome [79].
The field of genomic sequencing is undergoing a transformative shift, driven by relentless innovation in both short-read (SR) and long-read (LR) technologies. For researchers and drug development professionals, the contemporary landscape is no longer a binary choice but an expanding array of sophisticated tools. The decade-long dominance of a single sequencing paradigm is giving way to a more diverse and competitive market, where platforms are continuously redefined by breakthroughs in chemistry and engineering. Emerging challengers are accelerating progress, pushing the boundaries of read accuracy, throughput, and cost-effectiveness to unprecedented levels [104] [105].
This evolution is particularly critical for RNA sequencing, where the choice of technology directly impacts the ability to resolve complex transcriptional landscapes. While short-read platforms have set high standards for data quality and volume, long-read technologies have matured to offer high-fidelity (HiFi) accuracy and now address historical limitations in error rates [10]. The convergence is further evidenced by established long-read companies entering the short-read arena and vice-versa, fostering a period of intense innovation and cross-pollination that promises to redefine the capabilities of genomic analysis in research and clinical applications [105].
Platform & Chemistry Comparison Tables
To objectively compare the performance of modern sequencing platforms, the following tables summarize key specifications and representative experimental data from recent studies and technology assessments in 2024-2025.
Table 1: Sequencing Platform Specifications and Key Differentiators (2024-2025)
| Platform (Provider) | Technology Type | Key Chemistry/Chemistry Improvement | Representative Read Length | Claimed Accuracy (Phred Score) | Key Differentiator/Application Strength |
|---|---|---|---|---|---|
| NovaSeq X Series (Illumina) | Short-Read | Sequencing by Synthesis (SBS) | Short | Q30+ | High-throughput; large installed base; diverse application kits [105] |
| AVITI24 (Element Biosciences) | Short-Read | Sequencing by Binding (Avidity Cloudbreak) | Short | Q40+ | High accuracy; lower signal noise; cost-effective for high-throughput [104] [105] |
| UG 100 Solaris (Ultima Genomics) | Short-Read | Non-optical, sequencing on a wafer | Short | N/S | Ultra-low cost; claims the $80 genome [104] |
| Onso (PacBio) | Short-Read | Sequencing by Binding (SBB) | Short | Q40+ | High accuracy for variant calling; leverages PacBio's bioinformatics [10] [105] |
| Revio (PacBio) | Long-Read | Single Molecule Real-Time (SMRT) HiFi | 10-25 kb | Q30+ (HiFi) | High accuracy long reads; ideal for isoform sequencing & variant detection [10] |
| Sequel IIe (PacBio) | Long-Read | Single Molecule Real-Time (SMRT) | 10-25 kb | Q30+ (HiFi) | Foundational HiFi technology for full-length transcript sequencing [4] [37] |
| PromethION (Oxford Nanopore) | Long-Read | Nanopore-based electronic sensing | 5 kb - >1 Mb | ~Q28 | Ultra-long reads; real-time sequencing; direct RNA/DNA sequencing [105] |
| SBX System (Roche)* | Long-Read | Sequencing by Expansion (SBX); Nanopore | Mid-length | N/S | Novel chemistry creating "Xpandomers"; CMOS sensor detection [104] |
Note: *Roche SBX is announced for commercial release in 2026. N/S: Not Specified in search results.
Table 2: Comparative Performance in RNA Sequencing Applications from Recent Studies
| Experimental Metric | Illumina Short-Reads | PacBio Long-Reads (Iso-Seq) | Oxford Nanopore Long-Reads | Context & Notes |
|---|---|---|---|---|
| Throughput (Reads per cell) | Higher (~300,000 reads/cell) [4] | ~2M ZMW reads per SMRT cell [4] | Highest among long-read protocols (PCR-cDNA) [6] | Throughput influences depth of transcriptome coverage. |
| Transcript Coverage Uniformity | Bias at 5'/3' ends due to fragmentation [6] | Most uniform coverage across transcript length [6] | Higher 3' coverage (Direct RNA); uniform (PCR-cDNA) [6] | Affects quantitative accuracy across the entire transcript. |
| Full-Splice-Match Reads | Lower (inferred from read length) | Higher (enabled by full-length transcript sequencing) [6] | High for PCR-cDNA protocol [6] | Critical for accurate isoform identification and quantification. |
| Gene Expression Correlation | High correlation with long-reads, but affected by artefacts [4] | High correlation after filtering of artefacts [4] | High correlation with expected spike-in concentrations [6] | Both can robustly quantify gene-level expression. |
| Isoform Discovery | Limited by indirect inference | High (direct observation of full-length isoforms) [8] [37] | High (direct observation of full-length isoforms) [6] | Long-reads are transformative for discovering novel isoforms. |
| Identification of Artefacts | Limited ability | Enabled by full-length context (e.g., TSO contamination) [4] | Enabled by full-length context | Specific library prep (e.g., MAS-ISO-seq) allows artefact removal. |
Detailed Experimental Protocols
To illustrate how comparative data is generated, this section details key methodologies from recent, influential studies that directly benchmark sequencing platforms.
Protocol 1: Cross-Platform Single-Cell RNA Sequencing Comparison
This protocol is derived from a 2025 study that sequenced the same 10x Genomics cDNA library on both Illumina and PacBio platforms to enable a per-molecule comparison [4].
- 1. Sample Preparation: Patient-derived organoid cells (clear cell renal cell carcinoma) were used. Cells were resuspended and loaded onto a 10x Genomics Chromium chip to generate single-cell Gel Beads-in-emulsion (GEMs) using the Chromium Single Cell 3' Reagent Kits (v3.1 Chemistry Dual Index) [4].
- 2. cDNA Generation: Within GEMs, full-length cDNA was synthesized via reverse transcription. The cDNA from all cells was then amplified and cleaned up using SPRI beads [4].
- 3. Library Preparation & Sequencing:
- For Illumina Short-Read Sequencing: The amplified cDNA was enzymatically sheared to 200-300 bp. Illumina sequencing libraries were constructed with end repair, A-tailing, adapter ligation, and sample index PCR. Sequencing was performed on an Illumina NovaSeq 6000 for paired-end 28-91 bp reads, targeting ~300,000 reads per cell [4].
- For PacBio Long-Read Sequencing: The same cDNA (45 ng input) was used for MAS-ISO-seq library preparation. A key step involved PCR with a modified primer to incorporate a biotin tag, enabling removal of template-switching oligo (TSO) artefacts. The cDNA was then processed for directional assembly into long MAS arrays (10-15 kb). Sequencing was performed on a PacBio Sequel IIe system [4].
- 4. Data Analysis: Reads were matched by cell barcode and unique molecular identifier (UMI). Bioinformatic processing used platform-specific pipelines (e.g., PacBio's Iso-Seq pipeline) followed by cross-comparison of mapped reads and generated gene count matrices [4].
Protocol 2: Systematic Benchmarking of RNA-seq Protocols (SG-NEx Project)
This protocol outlines the comprehensive approach of the Singapore Nanopore Expression (SG-NEx) project, which benchmarked five RNA-seq protocols across seven human cell lines in 2025 [6].
- 1. Sample and Spike-in Design: Seven human cell lines (e.g., HCT116, HepG2, A549) were selected. For a subset of runs, spike-in RNAs with known concentrations (Sequins, ERCC, SIRVs) were added to provide a ground truth for quantification accuracy [6].
- 2. Multi-Protocol Library Preparation: Each cell line was sequenced with multiple replicates using:
- Illumina short-read cDNA sequencing (SR).
- Nanopore direct RNA sequencing (direct RNA).
- Nanopore amplification-free direct cDNA sequencing (direct cDNA).
- Nanopore PCR-amplified cDNA sequencing (cDNA).
- PacBio IsoSeq (IsoSeq) [6].
- 3. Unified Data Processing: Data from all protocols were processed through a standardized, community-curated pipeline (
nf-core/nanoseq). This pipeline performs quality control, alignment, transcript discovery and quantification, and differential expression analysis, ensuring a fair comparison [6]. - 4. Performance Metric Analysis: The study compared protocols based on throughput, read length, transcript coverage uniformity, gene/transcript expression correlation with spike-ins and between protocols, and the ability to identify alternative isoforms and fusion transcripts [6].
Technology Selection Workflow
The following diagram illustrates a decision-making workflow for selecting a sequencing technology based on common research objectives, integrating findings from the cited comparisons.
Diagram 1: A workflow for selecting a sequencing technology based on primary research objectives, highlighting the strengths of different platforms.
The Scientist's Toolkit: Key Research Reagent Solutions
Critical experimental outcomes depend on the choice of foundational reagents and technologies. The following table details key solutions referenced in the featured experimental protocols.
Table 3: Essential Research Reagents and Platforms for Sequencing Studies
| Item/Solution | Function in Research | Example Use-Case |
|---|---|---|
| 10x Genomics Chromium | Partitions single cells into GEMs for barcoding RNA transcripts, enabling single-cell resolution. | Preparing single-cell cDNA libraries from heterogeneous tissue or cell cultures for downstream sequencing on any platform [4]. |
| PacBio MAS-ISO-seq Kit | Prepares long-read libraries from cDNA by concatenating transcripts, increasing throughput and enabling artefact removal. | Generating high-throughput Iso-Seq libraries from full-length cDNA for identifying novel isoforms on Sequel IIe or Revio systems [4]. |
| Spike-in RNA Controls (e.g., SIRV, ERCC) | Provides an internal standard with known concentration and sequence to benchmark quantification accuracy across protocols. | Objectively evaluating the performance and bias of different RNA-seq library prep and sequencing methods [6]. |
| nf-core/nanoseq Pipeline | A community-curated, standardized bioinformatics workflow for processing long-read RNA-seq data. | Ensuring reproducible alignment, quantification, and quality control of Nanopore or PacBio data in a containerized environment [6]. |
| SQANTI3 | A comprehensive tool for the quality control, classification, and curation of long-read transcripts. | Characterizing and filtering isoforms discovered by PacBio or Oxford Nanopore sequencing against a reference annotation [4] [37]. |
| GRCh38 & T2T-CHM13 | Reference genomes used for read alignment and annotation. GRCh38 is standard; T2T offers more complete sequences in repetitive regions. | Aligning sequencing reads for transcript discovery and quantification. T2T-CHM13 may improve analysis in previously unresolved genomic regions [37]. |
The future landscape of sequencing is characterized by specialization and convergence. No single platform universally outperforms all others; instead, the choice is increasingly dictated by the specific biological question. Short-read technologies continue to advance in accuracy and cost-reduction, solidifying their role in high-throughput, quantitative gene expression studies [105]. Concurrently, long-read technologies have overcome historical accuracy barriers and are now transformative for applications demanding isoform-resolution, such as in cancer research and the study of complex genetic disorders [8] [37].
The most powerful future approaches will likely leverage the complementary strengths of both technologies. The development of integrated analysis pipelines and benchmarked datasets, like those from the SG-NEx project, provides researchers with the tools to make informed decisions and implement robust, multi-platform strategies [6]. As chemistry improvements continue to emerge from both established leaders and new entrants, the potential for discovery in transcriptomics and drug development will only expand, making this a uniquely dynamic and promising era for genomic science.
Conclusion
Short-read and long-read RNA sequencing are not competing but complementary technologies that form a powerful toolkit for modern biomedical research. Short-read sequencing remains the workhorse for high-throughput, cost-effective gene expression quantification and variant detection, while long-read technologies are indispensable for unraveling transcriptomic complexity, including full-length isoform resolution, structural variant detection, and direct RNA modification analysis. The choice between them is dictated by the specific research goal, genome complexity, and available resources. For comprehensive insights, a hybrid approach that leverages the strengths of both is often most powerful. Future directions will see increased integration of these technologies into clinical pipelines, driven by continuous improvements in accuracy, cost, and analytical tools, ultimately accelerating personalized medicine and the development of novel therapeutics.
References
- 1. Long-read RNA sequencing: A transformative technology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11897757/]
- 2. Transcriptomics in the era of long-read sequencing [https://www.rna-seqblog.com/transcriptomics-in-the-era-of-long-read-sequencing/]
- 3. Sequencing 101: long-read sequencing [https://www.pacb.com/blog/long-read-sequencing/]
- 4. Comparison of single-cell long-read and short ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12231600/]
- 5. RNA-seq data science: From raw data to effective ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.997383/full]
- 6. A systematic benchmark of Nanopore long-read RNA ... [https://www.nature.com/articles/s41592-025-02623-4]
- 7. Methodological Comparison of Short-Read and Long ... [https://www.mdpi.com/1422-0067/26/18/9254]
- 8. Long-read RNA sequencing: A transformative technology ... [https://www.sciencedirect.com/science/article/pii/S1525001624007524]
- 9. Short read vs long read sequencing - INTEGRA Biosciences [https://www.integra-biosciences.com/united-states/en/blog/article/short-read-vs-long-read-sequencing]
- 10. Long-read sequencing vs short-read sequencing [https://frontlinegenomics.com/long-read-sequencing-vs-short-read-sequencing/]
- 11. NGS-Based RNA-Sequencing Market Size 2024 - 2034 [https://www.towardshealthcare.com/insights/ngs-based-rna-sequencing-market-sizing]
- 12. DNA Sequencing: How to Choose the Right Technology [https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/]
- 13. Comparing long-read sequencing technologies [https://www.pacb.com/blog/sequencing-101-comparing-long-read-sequencing-technologies/]
- 14. Benefits and Applications of Long-Read Sequencing [https://geneviatechnologies.com/blog/benefits-and-applications-of-long-read-sequencing/]
- 15. Powered by PacBio: Selected publications from August 2025 [https://www.pacb.com/blog/powered-by-pacbio-selected-publications-from-august-2025/]
- 16. Powered by PacBio: Selected publications from June 2025 [https://www.pacb.com/blog/powered-by-pacbio-selected-publications-from-june-2025/]
- 17. Advances in nanopore direct RNA sequencing and its ... [https://www.sciencedirect.com/science/article/abs/pii/S073497502500196X]
- 18. Comparative analysis of Illumina, PacBio, and nanopore ... [https://www.frontiersin.org/journals/microbiomes/articles/10.3389/frmbi.2025.1587712/full]
- 19. A comparative evaluation of computational models for RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346441/]
- 20. Accurate circular consensus long-read sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6776680/]
- 21. TopoQual polishes circular consensus sequencing data and ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-06020-0]
- 22. Benchmarking reveals superiority of deep learning variant ... [https://elifesciences.org/articles/98300]
- 23. Performance assessment of DNA sequencing platforms in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8985210/]
- 24. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 25. Short-Read Sequencing vs. Long- ... [https://www.zymoresearch.com/blogs/blog/short-read-vs-long-read-sequencing?srsltid=AfmBOopl6K8HdAt4JIzbo2UkGBmvAYd0tepb4FhK-6u4eIMOrH2Gk7Us]
- 26. The third generation sequencing: the advanced approach to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7237973/]
- 27. What are the disadvantages of third generation NGS? [https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-disadvantages-of-third-generation-ngs]
- 28. Unlocking the Potential of Metagenomics with the PacBio High - Fidelity ... [https://pubmed.ncbi.nlm.nih.gov/39770685/]
- 29. Integrating short-read and long-read single-cell RNA ... [https://pubmed.ncbi.nlm.nih.gov/40050124/]
- 30. What is the Difference Between Short and Long-read RNA- ... [https://www.cd-genomics.com/resource-short-and-long-read-rna-seq.html]
- 31. Comparison of RNA-Sequencing Methods for Degraded RNA [https://pubmed.ncbi.nlm.nih.gov/38892331/]
- 32. Commentary: a review of technical considerations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12522984/]
- 33. Systematic evaluation of single-cell RNA-seq analyses ... [https://www.sciencedirect.com/science/article/pii/S2090123224002108]
- 34. RNA Sequencing in Drug Discovery and Development [https://www.lexogen.com/blog/rna-sequencing-in-drug-discovery-and-development/]
- 35. The Role of Long-Reads in Transcript Isoform Discovery ... [https://www.admerahealth.com/blog/the-role-of-long-reads-in-transcript-isoform-discovery-and-rna-analysis]
- 36. Transcriptomics in the era of long-read sequencing [https://pubmed.ncbi.nlm.nih.gov/40155769/]
- 37. Evaluation of Long-Read RNA Sequencing Procedures for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12469794/]
- 38. Long-read sequencing reveals the RNA isoform repertoire of ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03724-1]
- 39. Systematic assessment of long-read RNA-seq methods for ... [https://www.nature.com/articles/s41592-024-02298-3]
- 40. Using long-read sequencing technology to explore the ... [https://www.novogene.com/us-en/resources/blog/using-long-read-sequencing-technology-to-explore-the-complex-structure-of-transcripts/]
- 41. Long reads decipher genomes and transcriptomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12047276/]
- 42. From bulk, single-cell to spatial RNA sequencing [https://www.nature.com/articles/s41368-021-00146-0]
- 43. Understanding Bulk and Single-Cell RNA Sequencing [https://www.cd-genomics.com/resource-bulk-rna-sequencing-vs-single-cell-rna-sequencing.html]
- 44. Single‐cell RNA sequencing in cancer research [https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-01874-1]
- 45. Cancer cell states and emergent properties of the dynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8494223/]
- 46. Advances in single-cell RNA sequencing and its ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-023-01494-6]
- 47. Applications of single-cell sequencing in cancer research [https://pmc.ncbi.nlm.nih.gov/articles/PMC8190846/]
- 48. High-throughput single-сell sequencing in cancer research [https://www.nature.com/articles/s41392-022-00990-4]
- 49. Single‐cell RNA sequencing technologies and applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 50. Single-cell RNA sequencing technologies and ... [https://www.nature.com/articles/s12276-018-0071-8]
- 51. Single Cell Sequencing and The Future of Drug Discovery [https://www.parsebiosciences.com/blog/single-cell-sequencing-and-the-future-of-drug-discovery/]
- 52. Long-read RNA sequencing dataset of human pancreatic ... [https://www.nature.com/articles/s41597-025-05939-0]
- 53. Whole Transcriptome vs 3' mRNA-Seq: How to Choose ... [https://www.lexogen.com/blog/choosing-between-whole-transcriptome-total-rna-seq-and-3-mrna-seq/]
- 54. Systematic benchmarking of high-throughput subcellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534522/]
- 55. Spatial omics advances for in situ RNA biology [https://www.sciencedirect.com/science/article/pii/S1097276524006567]
- 56. Enhancing RNA Capture Efficiency in Spatial Transcriptomics [https://www.mdpi.com/1422-0067/26/22/11076]
- 57. Fast analysis of Spatial Transcriptomics (FaST) [https://pmc.ncbi.nlm.nih.gov/articles/PMC12004221/]
- 58. Development and application of a whole transcriptome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12063385/]
- 59. Expedited and accurate detection of targeted gene fusions ... [https://www.stjude.org/research/progress/progress-pulse/2025/expedited-and-accurate-detection-of-targeted-gene-fusions-with-stjude-fuzzion2.html]
- 60. Comprehensive profiling of RNA modification-related ... [https://www.nature.com/articles/s12276-025-01531-z]
- 61. A comparative evaluation of computational models for RNA ... [https://pubmed.ncbi.nlm.nih.gov/40802798/]
- 62. Bias in RNA-seq Library Preparation: Current Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8079181/]
- 63. Sample Preparation for NGS – A Comprehensive Guide [https://frontlinegenomics.com/sample-preparation-for-ngs-a-comprehensive-guide/]
- 64. Systematic evaluation of RNA-Seq preparation protocol ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5953-1]
- 65. Sequencing 101: How multiplexing with long-read ... [https://www.pacb.com/blog/sequencing-101-how-multiplexing-with-long-read-sequencing-makes-genomic-research-faster-cheaper-and-smarter/]
- 66. Experimental and Data Analysis Planning for RNA-Seq [https://www.lexogen.com/blog/rna-lexicon-experimental-and-data-analysis-planning-for-rna-sequencing/]
- 67. Comparative analysis of library preparation approaches for ... [https://www.nature.com/articles/s41598-025-12992-7]
- 68. Efficient experimental design and analysis strategies for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3560154/]
- 69. RNA-seq: impact of RNA degradation on transcript quantification [https://bmcbiol.biomedcentral.com/articles/10.1186/1741-7007-12-42]
- 70. Pervasive effects of RNA degradation on Nanopore direct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10251640/]
- 71. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 72. Modeling RNA degradation for RNA-Seq with applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC3616752/]
- 73. RNA-seq Data: Challenges in and Recommendations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4230301/]
- 74. Optimizing Budget and Resolution: Cost-Benefit Analysis ... [https://www.admerahealth.com/blog/optimizing-budget-and-resolution-costbenefit-analysis-of-short-vs-long-reads]
- 75. Long-Read Sequencing vs Short-Read Sequencing - Base4 [https://www.base4.co.uk/long-read-sequencing-vs-short-read-sequencing-a-comparison/]
- 76. Budgeting for an mRNA-seq project? How much does RNA ... [https://alitheagenomics.com/blog/budgeting-for-an-mrna-seq-project-here-are-the-main-cost-drivers-to-keep-an-eye-on]
- 77. A cost-effective RNA sequencing protocol for large-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4381617/]
- 78. A systematic benchmark of Nanopore long-read RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11978509/]
- 79. Targeted DNA-seq and RNA-seq of Reference Samples ... [https://www.nature.com/articles/s41597-024-03741-y]
- 80. Short-read Sequencing Market Forecast 2025 to 2035 [https://www.futuremarketinsights.com/reports/short-read-sequencing-market]
- 81. A Hitchhiker's Guide to long-read genomic analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12047252/]
- 82. Top 10 Bioinformatics Tools for scRNA-seq in 2025 [https://neovarsity.org/blogs/top-bioinformatics-tools-for-scrna-seq]
- 83. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 84. Top NGS Data Analysis Tools of 2025 [https://www.dromicsedu.com/blog-details/revolutionizing-genomics-top-ngs-data-analysis-tools-of-2025]
- 85. Performance evaluation of six popular short-read simulators [https://www.nature.com/articles/s41437-022-00577-3]
- 86. Benchmarking challenging small variants with linked and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9706577/]
- 87. Ultra-broad hybrid capture-based targeted next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576977/]
- 88. Comparison of RNA-Based Next-Generation Sequencing ... [https://pubmed.ncbi.nlm.nih.gov/34756276/]
- 89. A Hitchhiker Guide to Structural Variant Calling [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383524/]
- 90. A real-world multi-center RNA-seq benchmarking study ... [https://www.nature.com/articles/s41467-024-50420-y]
- 91. Benchmarking long-read RNA-sequencing analysis tools ... [https://www.nature.com/articles/s41592-023-02026-3]
- 92. Benchmarking long-read structural variant calling tools and ... [https://www.nature.com/articles/s41598-025-92750-x]
- 93. Benchmarking RNA-Seq quantification tools - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5003039/]
- 94. Blueprint Genetics' approach to pseudogenes and other ... [https://www.blueprintgenetics.com/pseudogene/]
- 95. Long-Read Sequencing and Structural Variant Detection [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293859/]
- 96. Complex genetic variation in nearly complete human ... [https://www.nature.com/articles/s41586-025-09140-6]
- 97. The Most Complete View of the Human Genome Yet Sets New ... [https://today.uconn.edu/2025/07/the-most-complete-view-of-the-human-genome-yet-sets-new-standard-for-use-in-precision-medicine/]
- 98. Illumina constellation mapped read technology uncovers ... [https://www.illumina.com/company/news-center/press-releases/2025/64dd710d-c79f-4b3a-be49-f3be33dff021.html]
- 99. How Kinnex long-read RNA sequencing wins out over ... [https://www.pacb.com/blog/publication-spotlight-how-kinnex-long-read-rna-sequencing-wins-out-over-illumina-for-transcript-level-quantification/]
- 100. To cut a short story long : development of full-length RNA - sequencing ... [https://era.ed.ac.uk/handle/1842/43495]
- 101. Singapore scientists unveil one of world’s largest long - read ... rna [https://www.a-star.edu.sg/gis/home/press-releases/press-releases-2025/singapore-scientists-unveil-one-of-world-s-largest-long-read-rna-sequencing-datasets-to-advance-disease-research]
- 102. Genomic data in the All of Us Research Program [https://www.nature.com/articles/s41586-023-06957-x]
- 103. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 104. The Best of NGS: Instrument Companies to Watch in 2025 [https://www.genengnews.com/topics/omics/the-best-of-ngs-instrument-companies-to-watch-in-2025/]
- 105. The Latest Developments in Sequencing Technologies [https://frontlinegenomics.com/the-latest-developments-in-sequencing-technologies/]