Toehold Switches for Viral RNA Detection: A Comprehensive Guide from Design to Diagnostic Application
This article provides a comprehensive overview of toehold switch technology for the detection of viral RNA, tailored for researchers and professionals in drug development.
Toehold Switches for Viral RNA Detection: A Comprehensive Guide from Design to Diagnostic Application
Abstract
This article provides a comprehensive overview of toehold switch technology for the detection of viral RNA, tailored for researchers and professionals in drug development. It covers the foundational principles of these synthetic riboregulators, which activate gene expression upon binding to a specific RNA trigger, sequestering the ribosome binding site until target recognition. The scope extends to detailed methodological protocols for designing and applying these biosensors for viruses such as SARS-CoV-2, Zika, and TuMV, often coupled with isothermal amplification. It further addresses critical troubleshooting and optimization strategies to enhance sensitivity and specificity. Finally, the article presents validation frameworks and comparative analyses with other diagnostic methods, highlighting the platform's potential for developing rapid, low-cost, and field-deployable diagnostics.
De Novo Design: Unraveling the Core Principles of Toehold Switch Technology
Toehold switches are a class of de-novo-designed prokaryotic riboregulators that activate gene expression in response to cognate RNAs with arbitrary sequences [1]. These synthetic RNA molecules employ toehold-mediated strand displacement (TMSD), a mechanism inspired by dynamic DNA nanotechnology, to provide precise translational control with high dynamic range and orthogonality [2] [1]. Unlike natural riboregulators that often rely on loop-loop interactions, toehold switches utilize linear-linear interactions initiated through single-stranded "toehold" domains, enabling more favorable reaction kinetics and greater design flexibility [1].
The significance of toehold switches extends across synthetic biology, molecular diagnostics, and therapeutic development. Their programmable nature allows researchers to construct complex genetic circuits, develop sensitive biosensors for pathogen detection, and implement sophisticated control systems for metabolic engineering [3] [1] [4]. The fundamental advantage of toehold switches lies in their ability to be forward-engineered to recognize virtually any RNA sequence, with demonstrated dynamic ranges often exceeding 400-fold between OFF and ON states [1].
Mechanism of Action
Fundamental Operating Principle
Toehold switches function through a conformational change triggered by specific RNA interactions. In their OFF state, the switch maintains a stable hairpin structure that sequesters the ribosome binding site (RBS) and start codon, preventing translation initiation [2] [1]. The switch transitions to the ON state when a trigger RNA molecule binds to the single-stranded toehold region (typically 12-18 nucleotides) and initiates strand displacement through branch migration, unwinding the inhibitory hairpin and exposing the RBS for ribosomal access [2] [3].
This mechanism differs fundamentally from natural riboswitches, which typically employ metabolite-binding aptamer domains to regulate gene expression. Instead, toehold switches are entirely RNA-programmable, with their trigger specificity determined by Watson-Crick base pairing rules [2]. The switching process is conceptually similar to computational strand displacement systems but operates within cellular environments.
Structural Components and Design Considerations
The architecture of a canonical toehold switch consists of several key structural elements:
- Toehold Domain: A single-stranded region (12-18 nt) that serves as the initial binding site for the trigger RNA [3]
- Stem Region: A double-stranded helix (12-16 bp) that sequesters the RBS and start codon in the OFF state [2]
- Loop Region: Contains the sequestered anti-RBS or anti-anti-RBS sequences and plays a crucial role in the structural transition [2]
- Expression Platform: The downstream coding sequence whose translation is regulated [2]
Table 1: Key Structural Components of a Basic Toehold Switch
| Component | Length | Function | Design Considerations |
|---|---|---|---|
| Toehold domain | 12-18 nt | Initiate trigger binding | Sequence complementary to trigger RNA |
| Stem region | 12-16 bp | Sequester RBS in OFF state | Thermodynamic stability critical for low leakage |
| Loop region | ~11 nt | Contain regulatory sequences | Avoid stacking interactions that impede switching |
| RBS/start codon | - | Translation initiation | Exposed upon trigger binding |
Recent designs have incorporated riboswitch-inspired elements, combining TMSD with switching principles from natural transcriptional and translational riboswitches [2]. Advanced implementations can regulate both translation and transcription, with some designs interfering with Rho-dependent termination or intrinsic terminators [2].
Applications in Viral RNA Detection
Toehold switches have emerged as powerful tools for diagnostic applications, particularly for detecting viral RNA pathogens. Their high specificity and programmability make them ideal for developing rapid, inexpensive point-of-care diagnostic platforms that can detect viral RNA without upstream amplification [3] [4].
SARS-CoV-2 Detection Platform
During the COVID-19 pandemic, toehold switches were engineered to detect SARS-CoV-2 RNA with high sensitivity and specificity. Researchers designed switches targeting conserved regions of the viral genome, particularly focusing on the nonstructural protein 2 (Nsp2) coding region [3]. Key design strategies included:
- Avoiding regions with high mutation rates or potential secondary structure
- Ensuring specificity against other human coronaviruses (HCoV-OC43, HCoV-229E, HCoV-NL63)
- Selecting genomic regions with minimal secondary structure to facilitate trigger binding
The most effective designs demonstrated sensitivity in the low picomolar range for direct target RNA detection, which could be enhanced to the low femtomolar range through signal amplification strategies [3].
Signal Amplification Strategies
To achieve clinically relevant sensitivity without target amplification, researchers have developed innovative signal amplification methods:
- TEV Protease Amplification System: Replacement of fluorescent reporters with tobacco etch virus (TEV) protease, which cleaves multiple quenched fluorescent substrates, significantly amplifying the detection signal [3]
- Coupled Transcriptional-Translational Control: Combining toehold switches with transcriptional regulators to enhance dynamic range [2]
- Orthogonal Switches: Using multiple toehold switches with minimal crosstalk for multiplexed detection [1]
Table 2: Performance Characteristics of Toehold Switch-based Viral Detection Systems
| Detection Platform | Target | Sensitivity | Amplification Method | Dynamic Range |
|---|---|---|---|---|
| Basic toehold switch | SARS-CoV-2 RNA | Low picomolar | None | ~400-fold |
| TEV-amplified system | SARS-CoV-2 RNA | Low femtomolar | Protease cleavage | Significantly enhanced |
| Cell-free paper-based | Various viral RNAs | Varies | NASBA/pre-amplification | >1000-fold |
Experimental Protocols
Toehold Switch Assembly and Cloning
This protocol describes the construction of toehold switch expression plasmids for bacterial systems, adapted from established methodologies [3] [5].
Materials Required
- Q5 High-Fidelity 2x Master Mix (NEB)
- T7 High Yield RNA Synthesis Kit (NEB)
- pET28a or similar expression vector
- DNA oligonucleotides for toehold switch and trigger
- Phusion High-Fidelity DNA Polymerase (ThermoScientific)
- T4 DNA ligase (ThermoScientific)
- Restriction enzymes (XbaI, BamHI, HindIII)
Procedure
Toehold Switch Design
- Design toehold switch sequence with 12-18 nt toehold domain and 18 nt stem sequence complementary to 30 nt trigger RNA
- Include GGG T7 promoter enhancer sequence at 5' end
- Design loop containing RBS followed by 18 nt complementary to first stem sequence with start codon 6 nt after loop
- Verify secondary structure using NUPACK or RNAfold
Plasmid Construction
- Synthesize toehold switch as 117 nt ultramer DNA oligonucleotide with XbaI site and 19 nt complementarity to reporter gene
- Amplify via PCR using Herculase II or Phusion high-fidelity polymerases
- Clone into expression vector (e.g., pUC19-mNeonGreen) using XbaI and BamHI sites
- Verify constructs by Sanger sequencing
Trigger RNA Construction
- Design 30 nt trigger sequence flanked by stem-loop structures at 5' and 3' ends
- Clone into appropriate expression vector or synthesize in vitro
Cell-Free Expression and Testing
Cell-free protein synthesis (CFPS) systems provide a rapid method for testing toehold switch performance without cellular transformation.
Materials Required
- PURExpress or similar cell-free transcription-translation system
- Fluorescent reporter (mNeonGreen preferred over GFP for enhanced signal)
- Synthetic trigger RNA
- Microplate reader for fluorescence detection
Procedure
Reaction Setup
- Combine CFPS components according to manufacturer instructions
- Add toehold switch plasmid (10-20 nM final concentration)
- Add trigger RNA in concentration series (0 pM to 1000 pM)
- Include no-trigger control for background measurement
Incubation and Measurement
- Incubate at 37°C for 4-8 hours
- Measure fluorescence hourly (mNeonGreen: Ex/Em ~506/517 nm)
- Calculate ON/OFF ratios as fluorescence with trigger divided by no-trigger control
Data Analysis
- Plot fluorescence versus trigger concentration to determine dynamic range
- Calculate EC50 from dose-response curve
- Assess leakiness from no-trigger control
Computational Design and Optimization
The design of high-performance toehold switches has been revolutionized by deep learning approaches that predict functionality from sequence data [6]. Traditional computational tools based solely on thermodynamic parameters often show poor correlation (as low as 0.22) with experimental performance [6].
Deep Learning Frameworks
Two complementary deep learning architectures have been developed specifically for toehold switch optimization:
- STORM (Sequence-based Toehold Optimization and Redesign Model): A convolutional neural network (CNN) framework that excels at identifying important sequence motifs and structural features [6]
- NuSpeak (Nucleic-Acid Speech): A natural language processing (NLP) approach that treats nucleotide sequences as "words" to learn grammatical rules governing toehold function [6]
These models were trained on a dataset of 91,534 toehold switches with experimentally characterized ON/OFF ratios, enabling accurate prediction of switch performance [6].
Key Design Principles Revealed by Deep Learning
Analysis of high-performing toehold switches has identified critical sequence features:
- NUA Motif: Positions 22-24 frequently contain NUA (where N is any nucleotide), creating a three-nucleotide bulge opposite the start codon [6]
- Nucleotide Preferences: Uracil over-represented and guanine under-represented in positions immediately before the Shine-Dalgarno sequence in high-performing switches [6]
- Start Codon Context: Avoidance of in-frame stop codons in the descending stem region [6]
- Amino Acid Bias: Small hydrophobic amino acids (valine, alanine, glycine) are preferred at the N-terminus of the reporter protein [6]
Advanced Applications and Future Directions
Integration with CRISPR Systems
Recent work has demonstrated sophisticated integration of toehold switches with CRISPR-Cas systems, creating intelligent genetic circuits with enhanced functionality. The intelligent guide RNA (IngRNA) platform incorporates dual toehold switches that regulate Cas9 activity in response to specific trigger RNAs [7].
In this system:
- The CrRNA sequence is sequestered by flanking complementary sequences
- Trigger RNA binding to the first toehold initiates a cascade that releases functional CrRNA
- This enables conditional CRISPR activity dependent on cellular RNA triggers [7]
Such systems demonstrate the potential of toehold switches as components of complex genetic computers that can process intracellular information and execute programmed responses.
Research Reagent Solutions
Table 3: Essential Research Reagents for Toehold Switch Development
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Polymerases | Q5 High-Fidelity (NEB), Phusion (ThermoScientific), Herculase II (Agilent) | PCR amplification of toehold switch constructs |
| Cloning Systems | pET28a, pUC19, pTargetF, pX458 | Expression vectors for bacterial and mammalian systems |
| Cell-Free Systems | PURExpress (NEB), TX-TL | Rapid in vitro testing of switch performance |
| Reporter Genes | mNeonGreen, eGFP, luciferase, β-galactosidase | Quantitative assessment of translation activation |
| Computational Tools | NUPACK, ViennaRNA, STORM, NuSpeak | Prediction and optimization of switch designs |
Toehold switches represent a versatile and powerful platform for translational control with significant applications in viral detection and synthetic biology. Their programmable nature, high dynamic range, and orthogonality make them ideal components for diagnostic systems and genetic circuits. Current research continues to expand their capabilities through integration with amplification strategies, CRISPR systems, and deep learning-based design tools. As computational design methods improve and our understanding of RNA structure-function relationships deepens, toehold switches are poised to become increasingly sophisticated tools for biomedical research and clinical applications.
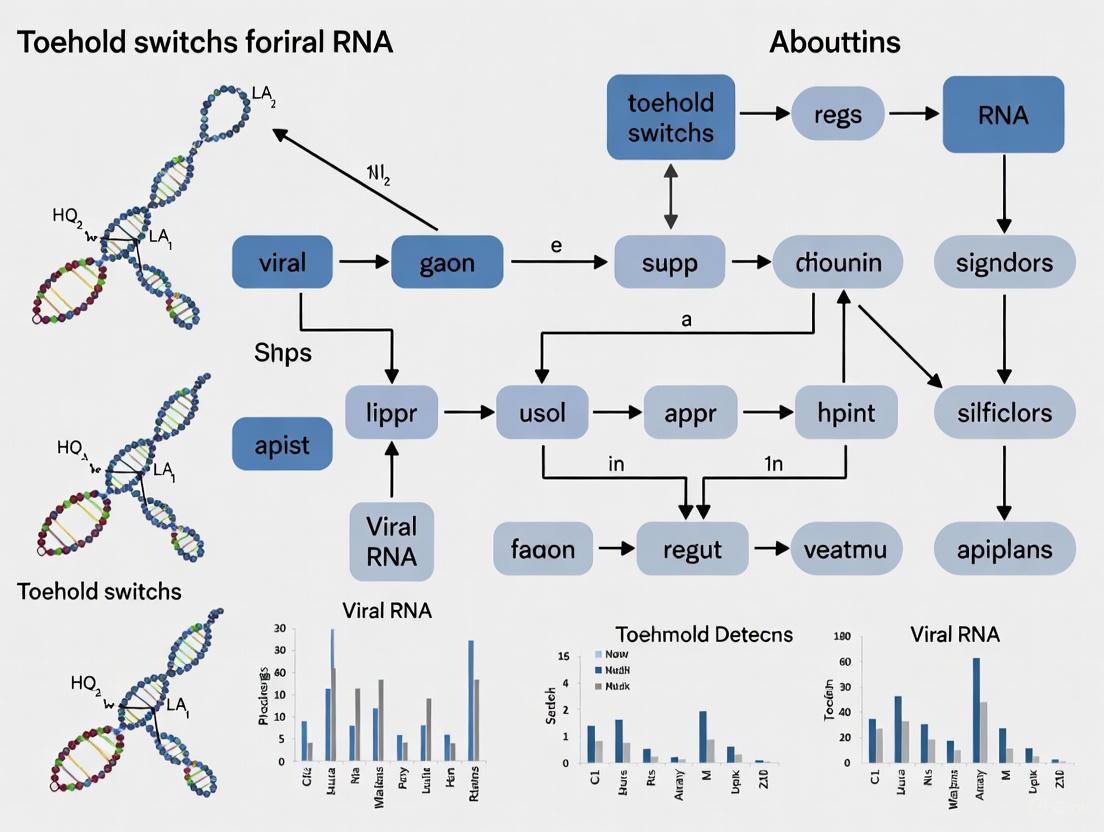
Toehold switches are synthetic RNA molecules that act as precise "on/off" switches for gene expression, specifically engineered to detect the presence of a target RNA sequence. In their inactive state, the toehold switch folds into a hairpin secondary structure that physically blocks the ribosome binding site (RBS) and the start codon (AUG), thereby preventing the initiation of translation and the synthesis of a reporter protein [3] [8]. Detection occurs via toehold-mediated strand displacement: a specific RNA "trigger" from a target (e.g., a viral genome) binds to a complementary single-stranded "toehold" region on the switch. This binding initiates a cascade of structural changes that ultimately exposes the RBS, allowing the ribosome to bind and initiate translation of a downstream reporter gene [3] [7] [9]. This mechanism provides a powerful, programmable tool for viral RNA detection without the need for complex protein-based sensors.
Detailed Mechanism Breakdown
The activation process can be broken down into three distinct stages, as illustrated in the diagram below.
Toehold Binding
The process is initiated when the single-stranded toehold region (typically 10-15 nucleotides) of the switch recognizes and binds to a complementary sequence on the trigger RNA via Watson-Crick base pairing [3] [9]. This initial interaction is highly specific and does not require energy input, as the binding is driven by the thermodynamic drive to maximize base pairing. The length and sequence of the toehold can be tuned to optimize both the specificity and the binding kinetics of the sensor.
Strand Displacement
The binding to the toehold region creates a three-stranded intermediate. The trigger RNA then proceeds to "invade" the double-stranded stem of the hairpin through a process called branch migration [9]. In this step, the trigger strand systematically displaces the incumbent strand that forms the stem, base by base. The energy gained from forming new base pairs between the trigger and the stem destabilizes and unwinds the original hairpin structure. Single-molecule force spectroscopy studies have revealed that this invasion process can proceed very rapidly, with single step times on the order of microseconds [9].
RBS Exposure
The successful strand displacement causes a major conformational change in the toehold switch, disrupting the stem-loop that was sequestering the RBS and start codon [3] [10]. With these key translational initiation elements now exposed and accessible, the ribosome can bind, initiating the translation of the downstream reporter protein. The reporter, such as a fluorescent protein (e.g., eGFP, mNeonGreen) or an enzyme (e.g., LacZ, TEV protease), generates a quantifiable signal indicating a positive detection event [3] [10].
Performance Metrics and Quantitative Data
The performance of toehold switches is quantified by their sensitivity, specificity, and dynamic range. The following table summarizes key performance data from recent research, highlighting how different design strategies impact the limit of detection (LOD).
Table 1: Performance Metrics of Toehold Switch-Based Sensors
| Target Analyte | Reporter System | Key Design Feature | Limit of Detection (LOD) | Reference / Context |
|---|---|---|---|---|
| SARS-CoV-2 RNA | mNeonGreen | Optimized switch design (CSU 08) | Low picomolar (pM) range | [3] |
| SARS-CoV-2 RNA | TEV protease + quenched fluorescent reporter | Downstream enzymatic signal amplification | Low femtomolar (fM) range | [3] |
| Turnip Mosaic Virus (TuMV) RNA | LacZ (colorimetric) | Coupled with NASBA pre-amplification | <10 fM | [11] |
| General Toehold Switch | Luciferase | Dual toehold switches (IngRNA platform) | High ON/OFF ratios reported | [7] |
Detailed Experimental Protocol
This protocol details the key steps for testing a toehold switch sensor in a cell-free protein synthesis (CFPS) system, a common platform for rapid diagnostic development [3].
Protocol: Testing Toehold Switch Activation in a Cell-Free System
I. Principle The functionality of a designed toehold switch is validated by incubating its DNA template in a CFPS reaction. The addition of a synthetic trigger RNA should activate the switch, leading to the production of a reporter protein, the signal of which is measured and compared to a no-trigger control.
II. Reagents and Equipment
- Toehold Switch DNA Template: Plasmid DNA or a PCR product containing a T7 promoter, the toehold switch sequence, and the reporter gene open reading frame [3].
- Synthetic Trigger RNA: A single-stranded RNA oligonucleotide perfectly complementary to the toehold and stem region of the switch [3].
- Cell-Free Protein Synthesis System: A commercial or homemade extract (e.g., E. coli S30 extract) capable of transcription and translation [3].
- Negative Control: Nuclease-free water or a non-complementary RNA sequence.
- Microplate Reader or Fluorometer: For quantifying fluorescent or colorimetric reporter signal.
III. Procedure
- Reaction Setup: Prepare two 15 μL CFPS reactions on ice.
- Test Reaction: Contains CFPS mix, toehold switch DNA template (e.g., 10 nM), and synthetic trigger RNA (concentration to be determined by titration, e.g., 0.1-100 nM).
- Negative Control: Contains CFPS mix, toehold switch DNA template, and an equivalent volume of nuclease-free water.
- Incubation: Incubate both reactions at 37°C for 1-4 hours to allow for transcription, translation, and switch activation.
- Signal Measurement: After incubation, transfer the reactions to an appropriate plate or cuvette.
- For fluorescent reporters (e.g., mNeonGreen, eGFP): Measure fluorescence using the appropriate excitation/emission wavelengths (e.g., mNeonGreen: Ex/Em ~506/517 nm) [3].
- For colorimetric reporters (e.g., LacZ): Add a substrate like Chlorophenol Red-β-D-galactopyranoside (CPRG) and measure the absorbance shift at ~574 nm [11].
- Data Analysis: Calculate the fold change in signal by dividing the signal from the Test Reaction by the signal from the Negative Control. A successful switch will show a significant fold change (e.g., >10x) upon trigger addition.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials required for the development and testing of toehold switch-based biosensors.
Table 2: Essential Research Reagents for Toehold Switch Experiments
| Reagent/Material | Function/Description | Example Use Case |
|---|---|---|
| Toehold Switch Plasmid | Vector containing T7 promoter, toehold switch sequence, and reporter gene (e.g., mNeonGreen, LacZ). | Serves as the DNA template for in vitro transcription/translation or for transfection into cells [3] [10]. |
| Trigger RNA | Synthetic single-stranded RNA oligonucleotide complementary to the toehold switch's target site. | Used as a positive control to validate switch function and for sensitivity assays [3]. |
| Cell-Free Protein Synthesis (CFPS) System | A crude cellular extract (e.g., from E. coli) supplying the machinery for transcription and translation. | Provides a rapid, cell-free environment for testing switch activity and developing diagnostics [3] [11]. |
| Lipofectamine | A lipid-based transfection reagent. | Used to deliver toehold switch plasmids into mammalian cells for validation in a cellular context [10]. |
| T7 High Yield RNA Synthesis Kit | A commercial kit for in vitro transcription of RNA. | Used to synthesize large quantities of trigger RNA or the toehold switch RNA itself [7]. |
| Nucleic Acid Sequence-Based Amplification (NASBA) | An isothermal RNA amplification technique. | Pre-amplifies target viral RNA from samples to enhance detection sensitivity to clinically/filed-relevant levels (fM) [11]. |
Advanced Applications and Signal Amplification
To achieve the femtomolar sensitivity required for clinical or field diagnostics, the basic toehold switch mechanism is often integrated with signal amplification strategies. The workflow below illustrates two powerful approaches: upstream target amplification and downstream signal amplification.
Upstream Target Amplification (Left Pathway): Methods like Nucleic Acid Sequence-Based Amplification (NASBA) are used to directly amplify the target viral RNA from a sample before it interacts with the toehold switch. This generates a large number of trigger RNA molecules, enabling detection even from very low initial concentrations [11].
Downstream Signal Amplification (Right Pathway): Instead of amplifying the target, this strategy amplifies the output signal. The toehold switch is designed to control the expression of a highly active enzyme, such as Tobacco Etch Virus (TEV) protease. A single activated switch can produce multiple TEV protease molecules. Each protease molecule can then cleave many copies of a quenched fluorescent substrate, leading to a substantial amplification of the final detectable signal [3].
Toehold switches are synthetic riboregulators that have emerged as powerful tools in synthetic biology and diagnostics, enabling the programmable detection of specific RNA sequences. Their core function relies on a precise structural arrangement that transitions from a silent "OFF" state to an active "ON" state upon encountering a trigger RNA, making them particularly valuable for applications such as viral RNA detection [12] [13]. The operational elegance of the toehold switch stems from three interdependent structural components: the toehold domain that initiates target recognition, the stem-loop that structurally represses translation, and the sequestered start codon that prevents unintended protein synthesis. This architecture allows for the creation of highly specific and sensitive biosensors that can be deployed in both cell-free systems and within living cells to detect pathogen RNA, including that of coronaviruses such as SARS-CoV-2 [12] [14]. The design principles of these switches capitalize on predictable RNA-RNA interaction kinetics and strand displacement mechanisms, offering a versatile platform for engineering diagnostic tools and genetic circuits.
Detailed Structural and Functional Analysis
Toehold Domain: The Recognition Module
The toehold domain is a single-stranded RNA region, typically 10-30 nucleotides in length, located at the 5' end of the switch. It serves as the initial binding site for the complementary trigger RNA through Watson-Crick base pairing [13]. This domain functions as a molecular catalyst that initiates a strand displacement reaction. The binding of the trigger RNA to the toehold domain is reversible and follows second-order kinetics, providing the switch with its programmable specificity. Once the trigger RNA successfully binds to the toehold, it nucleates a progressive zippering effect that propagates through the adjacent regions, ultimately unraveling the inhibitory secondary structure of the switch. The sequence and length of the toehold domain can be systematically optimized to balance binding affinity and specificity, minimizing off-target interactions while maintaining sensitivity to the intended target [7]. In viral detection applications, the toehold domain is designed to be perfectly complementary to a unique sequence within the viral genome, enabling precise pathogen identification.
Stem-Loop: The Structural Repressor
The stem-loop structure forms the central repressive element of the toehold switch, maintaining the system in its "OFF" state in the absence of the trigger RNA. This element typically consists of a double-stranded stem region (often with high GC content for stability) flanking a single-stranded loop [13]. The stem-loop serves two critical functions: first, it sterically blocks access to the ribosome binding site (RBS) and start codon; second, it provides the thermodynamic barrier that prevents spontaneous activation. The stability of this stem-loop is carefully balanced during design—too stable, and the switch cannot be efficiently activated by the trigger; too unstable, and the switch exhibits high background expression (leakiness) [12]. Computational tools like NUPACK and ViennaRNA are essential for predicting the folding and stability of this structural element, ensuring optimal switch performance [13] [14]. For example, in coronavirus detection platforms, stem lengths between 4-15 nucleotides and loop sizes of 3-10 nucleotides have been successfully employed [14].
Start Codon Sequestration: The Translational Block
Start codon sequestration represents the functional mechanism by which toehold switches control translation. In the OFF state, the start codon (AUG) and often the adjacent Shine-Dalgarno sequence (in prokaryotes) are embedded within the stable stem-loop structure, rendering them inaccessible to the translation initiation machinery [13] [14]. This sequestration physically prevents ribosome binding and scanning, thereby blocking translation of the downstream reporter or effector gene. Upon trigger RNA binding and subsequent strand displacement, the stem-loop unwinds, exposing the start codon and RBS to ribosomes. This structural transition activates translation, leading to the production of reporter proteins such as β-galactosidase, luciferase, or GFP, which provide a measurable signal indicating target detection [12] [14]. The precise positioning of the start codon within the stem is critical—it must be sufficiently buried to prevent leaky expression yet completely accessible upon switch activation to maximize signal output.
Table 1: Quantitative Design Parameters for Toehold Switch Components
| Structural Component | Key Parameters | Typical Range | Design Considerations |
|---|---|---|---|
| Toehold Domain | Length | 10-30 nt | Longer toeholds increase binding affinity but may reduce specificity |
| GC Content | 40-60% | Affects binding kinetics and melting temperature | |
| Location | 5' end of switch | Must be single-stranded and accessible for trigger binding | |
| Stem-Loop | Stem Length | 4-15 bp | Shorter stems reduce stability; longer stems increase activation energy |
| Loop Size | 3-10 nt | Affects structural flexibility and accessibility | |
| Minimum Free Energy | -15 to -30 kcal/mol | Determines switch stability and propensity for spontaneous opening | |
| Start Codon Region | Positioning | Within stem | Critical for effective sequestration in OFF state |
| Flanking Sequences | 5-8 nt upstream/downstream | Influences ribosome binding efficiency in ON state | |
| Context Sequence | AUG followed by optimal codons | Enhances translational efficiency upon activation |
Application in Viral RNA Detection
The structural components of toehold switches make them exceptionally well-suited for viral RNA detection, as demonstrated in multiple diagnostic platforms for coronaviruses including SARS-CoV-2 and MERS-CoV [14]. When designed to target conserved regions of viral genomes, the toehold domain provides sequence specificity, while the stem-loop and start codon sequestration mechanisms ensure minimal background signal in uninfected samples. The activation of the switch leads to the production of easily detectable reporter proteins, enabling colorimetric, fluorescent, or luminescent readouts.
In one implementation, toehold switches targeting SARS-CoV-2 were coupled with isothermal amplification methods like reverse transcription loop-mediated amplification (RT-LAMP) to enhance sensitivity. This integrated approach achieved detection limits as low as 120 copies of coronavirus RNA within 70 minutes, demonstrating the practical utility of these structural principles in clinical diagnostics [14]. The specificity inherent in the toehold domain design allowed discrimination between different coronaviruses, a critical requirement for accurate diagnosis. Furthermore, the modular nature of the toehold switch architecture permits rapid redesign to target emerging viral variants by simply modifying the toehold domain sequence while maintaining the same structural scaffold and detection methodology.
Experimental Protocol: Designing and Validating Toehold Switches for Viral RNA Detection
Computational Design and In Silico Optimization
Objective: To design a toehold switch specific to a target viral RNA sequence and predict its secondary structure and binding characteristics.
Procedure:
- Target Sequence Identification: Select a unique ∼30 nt target region within the viral genome (e.g., SARS-CoV-2 nucleocapsid gene) using alignment tools to ensure specificity [14].
- Toehold Switch Design:
- Design the toehold domain to be perfectly complementary to the target viral sequence.
- Incorporate a stem-loop structure with the start codon (AUG) embedded within the stem.
- Maintain a constant 5' and 3' scaffold sequence for proper folding and reporter gene compatibility.
- In Silico Folding Analysis:
- Use NUPACK software to model the secondary structure of the designed switch both with and without the trigger RNA [12] [14].
- Calculate the normalized ensemble defect (NED), selecting designs with NED < 10% for experimental testing [14].
- Verify that the start codon remains sequestered in the unbound state and accessible in the trigger-bound state.
- Specificity Check: Use BLAST to ensure the toehold domain does not have significant complementarity to non-target human transcripts.
Construction and In Vitro Testing
Objective: To experimentally validate the function and sensitivity of the designed toehold switch.
Materials:
- DNA oligonucleotides encoding the toehold switch and trigger sequence
- T7 High Yield RNA Synthesis Kit (for RNA transcription)
- Cell-free protein expression system (E. coli extract or commercial kit)
- Reporter plasmid with lacZ or GFP downstream of the switch
- Chlorophenol red-β-D-galactopyranoside (CPRG) for colorimetric assay if using lacZ
Procedure:
- Plasmid Construction:
- Clone the designed toehold switch sequence into a plasmid downstream of a T7 promoter and upstream of the reporter gene (e.g., lacZ) using Gibson assembly [12].
- Transform into E. coli DH5α and verify clones by Sanger sequencing.
- In Vitro Transcription-Translation:
- Set up cell-free reactions containing the toehold switch plasmid, T7 RNA polymerase, and translation machinery.
- Add trigger RNA at concentrations ranging from 0.1 nM to 500 nM to assess sensitivity [14].
- Incubate at 37°C for 4-6 hours to allow for expression.
- Signal Detection:
- For colorimetric detection with lacZ, add CPRG to the reaction and monitor the color change from yellow to purple at 570 nm [14].
- Calculate the fold-change between the triggered and untriggered states, selecting switches with >50-fold induction for further application.
Table 2: Troubleshooting Guide for Toehold Switch Validation
| Problem | Potential Cause | Solution |
|---|---|---|
| High background signal (leakiness) | Weak stem stability | Redesign stem with higher GC content or increased length |
| Low activation signal | Toehold domain inaccessible or stem too stable | Modify toehold length or redesign stem-loop structure |
| Poor specificity | Off-target trigger binding | Redesign toehold domain to reduce complementarity to non-target sequences |
| Inconsistent results | RNA degradation | Use RNase inhibitors and ensure proper RNA handling techniques |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Toehold Switch Development
| Reagent/Category | Specific Examples | Function in Toehold Switch Research |
|---|---|---|
| Design Software | NUPACK, ViennaRNA | Predicts secondary structure and facilitates switch design with minimal ensemble defect [13] [14] |
| Cell-Free Expression Systems | E. coli extracts, PURExpress | Provides in vitro environment for rapid switch validation without cellular complexity [12] |
| Reporter Systems | lacZ, GFP, luciferase | Quantifiable outputs for switch activation; each with different sensitivity and application suitability [12] [14] |
| Isothermal Amplification | RT-LAMP kits | Pre-amplification of viral RNA targets to enhance detection sensitivity for diagnostic applications [14] |
| RNA Production | T7 High Yield RNA Synthesis Kit | Generates high-quality trigger RNAs for validation and calibration [7] |
Structural and Operational Visualization
The following diagrams illustrate the key structural components and operational mechanism of toehold switches for viral RNA detection.
Diagram 1: Toehold Switch Structural Components
Diagram 2: Toehold Switch Operational Mechanism for Viral Detection
Diagram 3: Integrated Viral Detection Workflow
The precise integration of three structural components—the toehold domain, stem-loop, and sequestered start codon—enables toehold switches to function as highly specific and programmable biosensors for viral RNA detection. The structural principles outlined in this document provide a framework for designing and optimizing these synthetic riboregulators for diagnostic applications. When combined with amplification methods like RT-LAMP, toehold switch-based sensors achieve clinically relevant sensitivity for detecting pathogens such as SARS-CoV-2, offering rapid, colorimetric readouts suitable for point-of-care testing. The continued refinement of these structural components and their implementation in diagnostic platforms holds significant promise for addressing current and future infectious disease threats.
Toehold switches represent a class of de-novo-designed prokaryotic riboregulators that activate gene expression in response to cognate RNA triggers with arbitrary sequences [1]. These synthetic biological devices were developed to address a fundamental limitation in synthetic biology: the scarcity of composable, high-performance parts for constructing genetic circuits [1]. Unlike natural regulatory systems, which have evolved under multiple selective pressures, toehold switches are engineered from first principles using predictable Watson-Crick base pairing, enabling their programmability and expanding their dynamic range beyond natural systems [1] [15]. These riboregulators function through a unique mechanism where a trigger RNA binds to a toehold switch, causing a conformational change that exposes the ribosome binding site (RBS) and start codon, thereby activating translation of a downstream reporter gene [1] [11]. This mechanism differs significantly from natural riboregulators, which typically rely on loop-loop or loop-linear interactions and often bind directly to the RBS, imposing significant sequence constraints that limit their programmability and performance [1].
The architecture of toehold switches provides distinct advantages over natural systems. Traditional engineered riboregulators have typically demonstrated dynamic ranges up to approximately 55-fold for activators and 10-fold for repressors, whereas protein-based transcriptional regulators can achieve 350-480-fold modulation [1]. Toehold switches routinely achieve average dynamic ranges above 400-fold, matching or exceeding the performance of protein-based systems while offering greater programmability and design flexibility [1]. This performance, combined with their orthogonality and programmability, makes toehold switches particularly valuable for applications in molecular biology, synthetic biology, biotechnology, and diagnostic development [1] [16] [11].
Quantitative Advantages of Toehold Switches
The performance advantages of toehold switches over natural and earlier engineered systems can be quantified across several key parameters. The following table summarizes these comparative advantages based on experimental characterizations.
Table 1: Performance Comparison of Toehold Switches Versus Other Regulatory Systems
| System Type | Average Dynamic Range | Orthogonality (Number of Parts) | Crosstalk Level | Key Limitations |
|---|---|---|---|---|
| Natural Riboregulators | Varies; typically low | Limited by natural sequence constraints | Not systematically characterized | Evolved for specific biological contexts |
| Early Engineered Riboregulators | ~55-fold (activators); ~10-fold (repressors) | Libraries of up to 7 parts | ~20% crosstalk | Reliance on RBS binding and loop-mediated interactions |
| Protein-Based Transcriptional Regulators | 350-480-fold | Limited by available promoters/transcription factors | Variable | More difficult to program; larger genetic footprint |
| Toehold Switches | >400-fold (average); some individual switches >1000-fold | 26+ highly orthogonal systems demonstrated | <12% crosstalk in optimized sets | Performance depends on trigger accessibility and switch design |
The quantitative superiority of toehold switches stems from their innovative design principles. Unlike previous riboregulators that sequester the RBS to prevent translation, toehold switches sequester the region around the start codon while leaving the RBS accessible [1]. This design choice expands the sequence space available for programming and improves translational efficiency upon activation. Additionally, toehold switches employ linear-linear initiation domains rather than the loop-mediated interactions common in natural systems, resulting in more favorable reaction kinetics and thermodynamics [1]. The programmability of toehold switches is evidenced by their successful application in regulating 12 genes independently and constructing genetic circuits that compute 4-input AND logic, demonstrating their composability for complex synthetic biology applications [1].
Application Protocol: Viral RNA Detection Using Toehold Switches
This protocol details the application of toehold switches for detecting Turnip Mosaic Virus (TuMV) in Pseudostellaria heterophylla, as recently demonstrated by researchers [11]. The method combines nucleic acid sequence-based amplification (NASBA) with toehold switch activation in a cell-free system, enabling sensitive, specific, and equipment-free detection suitable for field applications.
Experimental Workflow
The following diagram illustrates the complete workflow for viral detection using toehold switch technology:
Step-by-Step Procedure
Toehold Switch Sensor Design and Screening
- Target Selection: Identify highly conserved regions within the viral genome. For TuMV, the HC-pro and CP regions are recommended due to their high conservation (mutation rates <12% in the protein sequence) [11].
- Sequence Analysis: Use computational tools such as MeFit Toehold Designer (https://github.com/mefit/toehold) and ViennaRNA (http://rna.tbi.univie.ac.at/) to predict minimum free energy secondary structures and thermodynamic parameters [11].
- Switch Design: Design toehold switches with the following characteristics:
- Free energies less than -20 kcal·mol⁻¹
- Trigger binding region complementary to 24-30 nt of the target viral sequence
- Conserved stem-loop structure that sequesters the start codon but not the RBS
- Sensor Screening: Test multiple switch candidates (typically 10-15 designs) against synthetic trigger RNAs to identify the highest-performing constructs before proceeding with viral detection experiments.
Sample Preparation and NASBA Amplification
- RNA Extraction: Extract total RNA from plant leaf tissue using either:
- Purified extraction methods (commercial kits) for maximum sensitivity
- Crude extraction protocols (grinding in buffer followed by centrifugation) for field applications
- NASBA Reaction Setup:
- Prepare the following reaction mixture:
- 5 μL of extracted RNA template
- 5 μL of 5× NASBA buffer (containing dNTPs, NTPs, and reaction salts)
- 2 μL of primer mix (10 μM each of forward and reverse primers targeting the viral sequence)
- 0.5 μL of enzyme mix (AMV reverse transcriptase, T7 RNA polymerase, and RNase H)
- 7.5 μL of nuclease-free water
- Incubate at 41°C for 90 minutes to achieve RNA amplification
- Heat-inactivate at 95°C for 5 minutes to stop the reaction
- Prepare the following reaction mixture:
Cell-Free Detection System
- Cell-Free Reaction Assembly:
- Combine the following components in a microcentrifuge tube:
- 5 μL of NASBA-amplified product
- 10 μL of cell-free extract (E. coli S30 or commercial cell-free system)
- 2 μL of toehold switch DNA construct (50 ng/μL)
- 2 μL of energy mix (containing ATP, GTP, CTP, UTP, and energy regeneration system)
- 1 μL of CPRG substrate (20 mM chlorophenol red-β-D-galactopyranoside)
- 5 μL of nuclease-free water
- Combine the following components in a microcentrifuge tube:
- Incubation and Signal Detection:
- Incubate the reaction at 37°C for 40-90 minutes
- Monitor color development visually or spectrophotometrically
- Positive samples develop a red color due to cleavage of the yellow CPRG substrate by β-galactosidase
- Result Interpretation:
- Strong red color: Positive detection (viral RNA present)
- No color change (yellow): Negative detection (viral RNA absent)
- Quantitative measurement: Measure absorbance at 574 nm for more precise quantification
Performance Metrics and Validation
Table 2: Performance Characteristics of Toehold Switch-Based TuMV Detection
| Parameter | Performance | Experimental Conditions |
|---|---|---|
| Detection Limit | 1 pM (40 min)10 fM (90 min) | With NASBA pre-amplification |
| Detection Time | 40-90 minutes (after NASBA) | Full protocol: ~3 hours |
| Specificity | No cross-reactivity with CMV | Tested against cucumber mosaic virus |
| Sample Type | Works with purified and crude RNA extracts | Suitable for field applications |
| Dynamic Range | >1000-fold in signal induction | From 10 fM to 1 nM target concentration |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of toehold switch technology requires specific reagents and components. The following table details the essential research tools and their functions.
Table 3: Essential Research Reagents for Toehold Switch Experiments
| Reagent/Category | Specific Examples | Function/Purpose |
|---|---|---|
| Design Tools | MeFit Toehold Designer, ViennaRNA, NUPACK | Predict RNA secondary structure, thermodynamics, and design optimal switch sequences |
| Cell-Free Systems | E. coli S30 extract, PURExpress | Provide transcriptional and translational machinery for in vitro testing |
| Reporter Systems | LacZ/CPRG, GFP/mut3b-GFP, Luciferase | Generate detectable signals (colorimetric, fluorescent) upon switch activation |
| Amplification Methods | NASBA, RPA, LAMP | Pre-amplify target RNA for enhanced sensitivity in detection applications |
| Vector Systems | pColE1, pColA, pET15b | Plasmid backbones for in vivo expression in bacterial systems |
| Experimental Strains | E. coli BL21 (DE3), DH5α | Model organisms for in vivo characterization and genetic circuit implementation |
Mechanism of Toehold Switch Operation
The molecular mechanism of toehold switches explains their superior performance characteristics compared to natural systems. The following diagram illustrates the structural transition and activation process:
This mechanism provides the foundation for the key advantages of toehold switches. The programmability stems from the arbitrary sequence recognition enabled by the toehold domain, which can be designed to complement any target RNA sequence without the constraints of natural riboregulator architectures [1]. The high dynamic range results from efficient sequestration of the start codon region in the OFF state and complete exposure in the ON state, combined with favorable linear-linear interaction kinetics that drive complete switching [1]. The orthogonality emerges from the vast sequence space available for designing multiple non-interacting switches, with demonstrated libraries of 26+ switches showing less than 12% crosstalk [1].
Recent advances in machine learning approaches have further enhanced toehold switch design and performance prediction. Deep learning models trained on large-scale datasets of toehold switch function (91,534 switches) have demonstrated significantly improved prediction accuracy (R² = 0.43-0.70) compared to traditional thermodynamic models (R² = 0.04-0.15) [15]. These computational approaches, combined with the experimental protocol outlined above, provide researchers with powerful tools for developing next-generation RNA detection systems and genetic circuits.
Toehold switches are a class of engineered riboregulators that provide programmable control of gene expression in response to specific RNA trigger sequences. These synthetic biology elements operate through a strand-displacement mechanism: in the absence of a trigger RNA, the switch folds into a stable hairpin structure that sequesters the ribosome binding site (RBS) and start codon, preventing translation. When a complementary trigger RNA binds to a single-stranded "toehold" region, it catalyzes the unfolding of the hairpin, exposing the RBS and start codon to initiate translation of a downstream reporter gene [13]. The predictable base-pairing rules of RNA make these switches highly amenable to computational design, yet their functional performance is heavily influenced by complex secondary structures that remain challenging to predict [12].
The rational design of high-performance toehold switches for viral RNA detection requires sophisticated in silico tools to model RNA secondary structure and hybridization dynamics. NUPACK and ViennaRNA represent two cornerstone software suites that enable researchers to analyze and design nucleic acid systems based on thermodynamic principles [13] [17]. These tools employ empirical free energy parameters derived from nearest-neighbor models to predict the minimum free energy (MFE) structures, equilibrium base-pairing probabilities, and hybridization behavior of RNA sequences [18] [19]. Within the context of viral detection research, these computational approaches help optimize switch sensitivity and specificity while minimizing leaky expression, ultimately accelerating the development of robust diagnostic platforms.
Computational Tools and Their Applications
NUPACK for Analysis and Design
NUPACK is a comprehensive software suite for analyzing and designing nucleic acid structures, devices, and systems. Its algorithms calculate the equilibrium base-pairing properties of complex ensembles of interacting nucleic acid strands, making it particularly valuable for modeling the multi-strand interactions between toehold switches and their viral RNA triggers [17]. The software supports various material types (RNA, DNA, or mixed) and can model different structural ensembles, including those with coaxial and dangle stacking, which are critical for accurate energy calculations [18].
For toehold switch design, NUPACK provides several key functionalities. The pairs and complexes commands can analyze the base-pairing probabilities between the switch and trigger RNA, while the concentrations command models the equilibrium concentrations of different complexes in solution. The platform's design algorithm focuses on minimizing the ensemble defect—the ensemble-averaged number of incorrectly paired nucleotides—to create sequences that robustly fold into desired structures [17]. When designing switches for viral detection, researchers can specify target complexes representing both the "OFF" state (switch alone) and "ON" state (switch bound to viral trigger), ensuring the switch remains off in the absence of the viral target while activating efficiently upon trigger binding.
Example NUPACK Model Configuration for Toehold Switches:
Example code snippet adapted from NUPACK documentation [18] [17]
ViennaRNA for Secondary Structure Prediction
The ViennaRNA package offers a complementary set of algorithms for RNA secondary structure prediction and analysis. Its core program, RNAfold, computes the minimum free energy secondary structure of a single RNA sequence, along with equilibrium base-pairing probabilities using partition function calculations [19]. Unlike NUPACK, which excels at modeling multi-strand interactions, ViennaRNA specializes in predicting the folding of individual RNA molecules, making it invaluable for ensuring that neither the toehold switch nor the viral trigger RNA contains stable internal structures that could interfere with their interaction [20].
For toehold switch design, ViennaRNA can predict and visualize the secondary structure of the switch in its unbound state, helping researchers identify and mitigate problematic structural features. The software can calculate the folding free energy, which correlates with switch performance—switches with excessively stable unbound structures may fail to activate, while those with insufficient stability may exhibit high background expression [20]. Recent benchmarking studies have indicated that ViennaRNA's predictions may show higher correlation with experimental data compared to other tools for certain RNA structures [20] [19].
Key ViennaRNA Commands for Toehold Switch Analysis:
Comparative Analysis of Prediction Accuracy
Recent large-scale experimental studies have quantified the performance of thermodynamic models compared to emerging deep learning approaches. A comprehensive analysis of 91,534 toehold switches revealed that while NUPACK and ViennaRNA provide valuable insights, their predictive power for actual switch function is limited.
Table 1: Performance Comparison of Toehold Switch Prediction Methods
| Prediction Method | R² for ON/OFF Ratio | Key Strengths | Key Limitations |
|---|---|---|---|
| NUPACK (MFE of RBS-linker) | 0.04 | Based on established thermodynamic principles | Poor correlation with experimental performance |
| NUPACK (IED) | 0.03 | Optimizes for ensemble defect in design | Limited predictive value for function |
| ViennaRNA (MFE) | Similar to NUPACK | Fast computation; user-friendly | Struggles with complex multi-state interactions |
| Deep Neural Networks | 0.43–0.70 | High predictive accuracy; pattern recognition | Requires large training datasets; less interpretable |
Data adapted from [15]
The relatively low correlation coefficients (R² = 0.04–0.15) for thermodynamic models highlight the challenges in predicting toehold switch function based solely on free energy calculations [15]. These models often fail to capture the complex kinetic and contextual factors that influence switch performance in biological systems, such as co-transcriptional folding effects and interactions with cellular components.
Integrated Design Protocol for Viral Detection Switches
Target Selection and Sequence Analysis
The design process begins with careful selection of target regions within the viral genome. For SARS-CoV-2 detection, researchers have successfully targeted conserved regions with low mutation rates, such as segments within the non-structural protein 2 (nsp2) coding region [3]. These regions should be analyzed for sequence conservation across viral variants and absence in human transcripts to ensure specificity. Tools like BLAST can identify unique viral sequences, while multiple sequence alignment reveals conserved regions.
Once a target region is identified, the complementary trigger sequence is designed. The ideal trigger length is typically 30-36 nucleotides, providing sufficient binding energy for efficient switch activation while maintaining specificity [3] [12]. The trigger sequence should be analyzed for internal secondary structure that might impede binding to the toehold switch. ViennaRNA's RNAfold can calculate the minimum free energy of the trigger alone, with lower (more negative) values indicating more stable internal structures that may reduce accessibility.
Toehold Switch Design and Optimization
The core toehold switch architecture follows established design principles with several key components:
- Toehold Domain: A single-stranded region (typically 12-15 nt) that initiates binding to the viral trigger RNA.
- Stem Region: A double-stranded segment that sequesters the RBS and start codon in the OFF state.
- Loop Region: Contains the sequestered RBS sequence.
- Linker Sequence: Connects the switch to the reporter gene [3] [20].
Table 2: Toehold Switch Design Parameters for Viral Detection
| Component | Optimal Length | Sequence Considerations | Design Tool |
|---|---|---|---|
| Toehold Domain | 12-15 nt | Fully complementary to viral target; avoid self-complementarity | NUPACK pairs |
| Stem Region | 18-21 bp per side | Moderate GC content (40-60%); avoid extreme stability | ViennaRNA RNAfold |
| Loop | 11 nt | Contains RBS (AGGAGA); fixed in B-series design | Fixed sequence |
| Start Codon | 3 nt (AUG) | Positioned in bulge region for proper sequestration | Structural analysis |
| Linker | 21-24 nt | Encodes low molecular weight amino acids; no secondary structure | Sequence optimization |
Parameters synthesized from [3] [12] [20]
The following workflow diagram illustrates the integrated computational design process for creating toehold switches targeting viral RNA:
Diagram 1: Computational design workflow for toehold switches. This integrated protocol combines target selection with iterative analysis using ViennaRNA and NUPACK to generate high-performance switches for viral RNA detection.
Using NUPACK, designers can model the interaction between the toehold switch and viral trigger. The complexes command calculates the free energy of hybridization, with more negative values indicating stronger binding. Additionally, the pairs command reveals the probability of base-pair formation at each position, helping identify regions of the switch that might misfold or form unintended structures. For the switch in its unbound state, ViennaRNA's RNAfold predicts the MFE structure and calculates its stability. Functional switches typically exhibit MFE values between -20 and -35 kcal/mol [20]. Switches with excessively stable structures (e.g., < -40 kcal/mol) may fail to activate, while those with insufficient stability (e.g., > -15 kcal/mol) may show high background expression.
Advanced Considerations for Viral Detection
Successful application of toehold switches for viral detection requires addressing several advanced design considerations:
Target Accessibility: The structural context of the target site within the viral genome significantly impacts switch performance. Research has shown that local base-pairing probabilities and secondary structure elements in the viral RNA can mask binding sites and hinder detection [12]. Tools like Toehold-VISTA integrate target structural features into machine learning models to improve design success rates [12].
Sequence Conservation: To ensure robust detection of evolving viral pathogens, toehold switches should target genomic regions with low mutation rates. For SARS-CoV-2, researchers have successfully designed switches against conserved regions in the nsp2 gene while avoiding mutational hotspots and structural protein genes under selective pressure [3].
Minimizing Off-Target Effects: Specificity is crucial for diagnostic applications. BLAST analysis against human transcripts can identify and eliminate designs with significant complementarity to endogenous RNAs. NUPACK's test tube analysis can model potential cross-reactions with abundant human RNAs.
Experimental Validation and Troubleshooting
In Vitro Characterization of Designed Switches
Computationally designed toehold switches require experimental validation to confirm function. The recommended approach involves in vitro testing using cell-free protein synthesis (CFPS) systems, which provide a controlled environment for initial characterization [3].
Protocol: Cell-Free Testing of Toehold Switches
- Plasmid Construction: Clone designed toehold switches upstream of a reporter gene (e.g., mNeonGreen, eGFP) in a vector containing a T7 promoter.
- In Vitro Transcription: Generate RNA triggers corresponding to the viral target sequence.
- CFPS Reaction: Combine purified switch plasmid (10-20 nM) with trigger RNA (0.1-1000 nM) in a commercial cell-free expression system.
- Fluorescence Measurement: Monitor reporter fluorescence over 4-16 hours at 37°C.
- Data Analysis: Calculate ON/OFF ratios by comparing fluorescence with and without trigger.
Well-performing switches typically show ON/OFF ratios >10:1, with detection sensitivity in the low picomolar range for the trigger RNA [3]. For SARS-CoV-2 detection, researchers have achieved sensitivity in the low femtomolar range by incorporating downstream signal amplification systems such as TEV protease cleavage cascades [3].
Troubleshooting Common Design Issues
Table 3: Troubleshooting Guide for Toehold Switch Design
| Problem | Potential Causes | Computational Diagnostics | Design Solutions |
|---|---|---|---|
| High Background (Leakiness) | Insufficient stem stability; alternative folding | Check MFE of switch alone (ViennaRNA); analyze alternative structures | Increase stem length; modify sequence to stabilize OFF state |
| Low Activation | Stable internal structure in trigger; switch too stable | Calculate MFE of trigger (ViennaRNA); check hybridization energy (NUPACK) | Redesign trigger target site; modify toehold length; decrease stem stability |
| Poor Specificity | Cross-hybridization with non-target sequences | BLAST against human transcriptome; NUPACK test tube analysis | Increase trigger length; modify toehold sequence for greater specificity |
| Inconsistent Performance | Co-transcriptional folding effects; kinetic traps | NUPACK partition function; analyze base-pair probabilities | Add 5' stabilizing hairpins; optimize nucleotide composition |
Troubleshooting guidance synthesized from [15] [3] [20]
The following experimental validation workflow outlines the key steps from computational design to functional confirmation:
Diagram 2: Experimental validation workflow. This process connects computational design with experimental testing, enabling iterative improvement of toehold switch performance for viral detection applications.
Research Reagent Solutions
Table 4: Essential Research Reagents for Toehold Switch Development
| Reagent/Category | Specifications | Function in Development | Examples/Notes |
|---|---|---|---|
| Nucleic Acid Design Tools | NUPACK 4.1; ViennaRNA 2.6.4 | Predict secondary structure; model hybridization | Free academic software; Python API available [18] [20] |
| Cell-Free Expression System | PURExpress; homemade extracts | In vitro testing of switch function | Commercial systems offer reproducibility [3] |
| Reporter Genes | mNeonGreen; eGFP; luciferase | Quantitative assessment of switch activity | mNeonGreen offers 5x intensity of eGFP [3] |
| Signal Amplification | TEV protease system | Enhance detection sensitivity | Enables femtomolar sensitivity without NASBA [3] |
| Vector System | pET; pUC derivatives | Switch and reporter expression | T7 promoter systems for high expression [3] [12] |
The rational design of toehold switches for viral RNA detection represents a powerful application of computational biology tools to address pressing diagnostic challenges. While NUPACK and ViennaRNA provide essential capabilities for modeling RNA secondary structure and hybridization dynamics, recent research indicates that thermodynamic parameters alone offer limited predictive power for actual switch function (R² = 0.04–0.15) [15]. The integration of these tools with emerging machine learning approaches, which demonstrate significantly higher correlation with experimental performance (R² = 0.43–0.70), represents the cutting edge of toehold switch design methodology [15] [12].
As the field advances, the most successful design pipelines will likely combine the mechanistic insights from thermodynamic modeling with the pattern recognition capabilities of deep learning. This hybrid approach promises to accelerate the development of highly sensitive and specific toehold switches for detecting viral pathogens, contributing to more responsive diagnostic platforms for emerging infectious diseases. For researchers embarking on toehold switch design, an iterative process that combines computational prediction with experimental validation remains essential for achieving robust performance in viral detection applications.
From Bench to Field: A Step-by-Step Guide to Building Toehold Switch Diagnostics
The rapid and specific detection of viral pathogens is a cornerstone of public health and diagnostic research. Among the most advanced tools emerging in this field are toehold switch riboregulators, programmable RNA sensors that offer high specificity and a direct visual readout. These synthetic biology elements are engineered to detect specific RNA sequences within a viral genome, initiating a molecular cascade that results in the production of a detectable signal, most commonly a fluorescent protein. This application note provides a detailed protocol for implementing a complete workflow, from the initial computational analysis of a viral genome to the final experimental validation using a visual reporter, specifically superfolder Green Fluorescent Protein (sfGFP). The entire process is framed within the context of detecting SARS-CoV-2 RNA, demonstrating a practical and critical application for modern viral diagnostics [12].
Workflow and Signaling Pathway
The entire process, from viral genome input to final fluorescent readout, involves a sequence of discrete molecular steps. The following diagram illustrates this integrated signaling pathway and experimental workflow.
Diagram 1: Toehold Switch Activation Pathway for Viral RNA Detection.
Research Reagent Solutions
Successful execution of this workflow requires a set of key reagents and molecular tools. The table below catalogs the essential components, their functions, and examples from the protocol.
| Reagent/Component | Function & Explanation | Example/Details |
|---|---|---|
| Toehold Switch Plasmid | Encodes the riboregulator; its transcription produces the sensor RNA that exists in an "OFF" state until triggered. | Second-generation design (tsgen2) with conserved stem to minimize variability [12]. |
| Target RNA Trigger | The viral RNA sequence that acts as the key; its binding to the toehold region initiates the strand displacement. | Can be in vitro transcribed or from extracted viral RNA (e.g., SARS-CoV-2) [12] [7]. |
| Reporter Plasmid (sfGFP) | Contains the reporter gene that is translated only upon switch activation, providing the visual signal. | sfGFP is preferred for its fast folding, high brightness, and improved signal-to-noise ratio [21]. |
| Cell-Free Expression System | A flexible, open platform for rapid testing; provides the transcriptional and translational machinery outside of a living cell. | Used for high-throughput screening and optimization of switch performance [12] [21]. |
| T7 High Yield RNA Synthesis Kit | For in vitro production of high-quality RNA molecules, including trigger RNAs for validation assays [7]. | Essential for generating defined trigger molecules to test sensor specificity [7]. |
| Two-Plasmid System | Allows for independent regulation and delivery of the sensor and trigger components within a single reaction or bacterial cell [12]. | pColA for switch expression and pET15b for target RNA expression [12]. |
Key Experimental Protocols
In Silico Target Site Selection and Sensor Design
Objective: To computationally identify accessible binding sites within the structured viral RNA genome and design specific toehold switches against them.
Methodology:
- Viral Sequence Acquisition: Obtain the complete RNA genome sequence of the target virus (e.g., SARS-CoV-2 from NCBI GenBank).
- Structural Accessibility Analysis: Use RNA folding software (e.g., NUPACK, ViennaRNA) to predict the secondary structure of the viral genome. Calculate local base-pairing probabilities to identify regions with low structural stability, which are more accessible for hybridization [12].
- Feature Extraction with Machine Learning: Integrate sequence-structure features (e.g., local MFE, codon usage bias, nucleotide content) into a machine learning framework like VISTA (versatile in-silico RNA-targeting analysis). Train a predictive model (e.g., Partial Least Squares Discriminant Analysis, PLS-DA) on high-throughput experimental data to capture the key determinants of sensor performance [12].
- Toehold Switch Design:
- Design a 30-36 nucleotide toehold switch trigger sequence that is fully complementary to the selected viral target site.
- Use established architectures (e.g., second-generation toehold switch) with a conserved stem-loop to minimize performance variability. The trigger RNA is designed to bind the toehold and unwind a specific number of base pairs (e.g., 6 bp) into the stem, initiating activation [12].
- Avoid introducing in-frame stop codons within the coding sequence of the switch itself.
Plasmid Construction and Molecular Cloning
Objective: To build the DNA templates necessary for expressing the toehold switch and the target RNA trigger.
Protocol for Switch and Reporter Plasmid Assembly:
- Template Design: Design DNA oligos for the toehold switch variant to include a 5' T7 promoter sequence, the switch sequence itself, a 21-nt linker, and the first 9 nt of the sfGFP reporter gene [12].
- Automated Cloning:
- Perform PCR amplification using high-fidelity DNA polymerase (e.g., Q5 Master Mix) with template DNA and specific primers [12].
- Digest the PCR product and the destination vector (e.g., pColADuet-1 for the switch) with appropriate restriction enzymes.
- Purify the digested fragments using a gel extraction kit.
- Assemble the insert and vector using Gibson assembly. Incubate the reaction at 50°C for 1 hour [12].
- Transform the assembled product into chemically competent E. coli DH5α and plate on LB agar with the appropriate antibiotic.
- Incubate overnight at 37°C.
- Purify validated plasmids using a miniprep kit for downstream applications [12].
Cell-Free Expression and Sensor Validation
Objective: To rapidly test the functionality, sensitivity, and specificity of the designed toehold switches in a controlled, cell-free environment.
Protocol for Cell-Free Testing:
- Reaction Setup: Reconstitute a cell-free transcription-translation (TXTL) system according to the manufacturer's instructions.
- Test Conditions:
- Experimental: Combine the toehold switch plasmid (e.g., 5-10 nM) with the cognate target RNA trigger (e.g., 10-100 nM).
- Negative Controls: Include reactions with switch plasmid alone (to measure leakiness), and with switch plasmid plus a non-cognate RNA trigger (to measure specificity).
- Positive Control: A plasmid with a constitutively expressed sfGFP can be used.
- Kinetic Measurement: Transfer the reaction mixture to a plate reader and monitor sfGFP fluorescence (Excitation: 488 nm, Emission: 509 nm) in real-time over 4-16 hours at 29-37°C [21].
- Data Analysis:
- Calculate the Fold Activation as the ratio of peak fluorescence in the experimental condition to the stable baseline fluorescence in the negative control (switch alone).
- Calculate the Signal-to-Noise Ratio by dividing the signal from the experimental well by the signal from the negative control.
- Perform statistical tests (e.g., t-test) to determine the significance (p-value) of the activation compared to controls [21].
Performance Data and Optimization
Quantitative data from iterative design-build-test-learn (DBTL) cycles is crucial for optimizing sensor performance. The table below summarizes key metrics from a representative optimization process, highlighting the impact of various design changes.
| Optimization Trial | Key Design Change | Peak sfGFP Output (a.u.) | OFF-State Baseline (a.u.) | Fold Activation | Specificity (p-value) |
|---|---|---|---|---|---|
| Trial 1 | AmilCP Chromoprotein | N/A (Colorimetric) | N/A (Colorimetric) | Confirmed Activation | 1.43 x 10⁻¹¹¹ [21] |
| Trial 2 | Standard GFP | ~400,000 | ~200,000 | ~2.0x | 5.00 x 10⁻³⁹ [21] |
| Trial 3 | Upstream Buffer Sequences | ~30,000 | ~25,000 | ~1.2x | 3.06 x 10⁻³¹ [21] |
| Trial 4 | Reduced Downstream G-Content | ~35,000 | ~25,000 | ~1.4x | 6.55 x 10⁻³⁸ [21] |
| Trial 5 | Superfolder GFP (sfGFP) | ~50,000 | ~25,000 | ~2.0x | 7.87 x 10⁻²⁵ [21] |
| Trial 10 | Final Validated Construct | ~70,000 | ~35,000 | ~2.0x | 7.42 x 10⁻³⁴ [21] |
Table 1: Quantitative Optimization of Toehold Switch Performance. Data adapted from cell-free expression experiments showing the progression of key performance metrics through iterative design cycles [21].
The following diagram summarizes the logical decision-making process and optimization strategies employed during the DBTL cycle to troubleshoot common issues like high background or low signal.
Diagram 2: Toehold Switch Optimization Decision Tree.
The emergence of rapidly mutating RNA viruses, such as SARS-CoV-2, has underscored the critical need for diagnostic and therapeutic strategies that remain effective across viral variants. A powerful approach to achieving this variant resilience is to target conserved, low-mutation regions within the viral genome. These regions are subject to strong evolutionary constraints, often because they are essential for viral replication or structural integrity, and thus accumulate fewer changes over time. For research focused on toehold switches—synthetic RNA sensors used for detection and gene regulation—targeting these conserved sequences ensures that the diagnostic and synthetic biology tools remain functional and specific even as the virus evolves. This application note details the bioinformatic and experimental protocols for identifying and validating these critical genomic regions, providing a robust framework for developing durable viral countermeasures.
Bioinformatics Workflow for Identifying Conserved Regions
Rationale and Strategic Approach
The genomic signature of a virus is shaped by evolutionary selection pressures to preserve sequences vital for its life cycle [22]. In the context of an emerging pathogen or a highly adapted virus, the available genomic data may exhibit low overall variability, making the distinction between conserved "signal" and variable "background" regions more challenging [23]. A successful strategy must therefore leverage all available information to pinpoint regions of unusually high conservation that cannot be explained solely by amino acid conservation, as these may indicate roles in RNA structure, packaging signals, or other non-coding functions [23].
A recommended five-step strategy for this process is outlined below [24]:
- Genome Segmentation: Divide the reference viral genome into short, overlapping oligonucleotides (k-mers, typically 20 nucleotides in length).
- Specificity Filtering: Discard k-mers that are not unique to the target virus. This includes sequences that appear more than once in the viral genome itself, or that are found in the host genome or in closely related species (e.g., common human coronaviruses).
- Conservation Analysis: Analyze the filtered k-mers against a large, global dataset of viral sequences (e.g., from GISAID) to identify those with the highest conservation rates across variants.
- Functional Filtering: Apply optimal assay design criteria (e.g., for qPCR or toehold switches) to the conserved k-mers, selecting those with appropriate GC content, melting temperature, and minimal secondary structure.
- Validation: Confirm the presence and specificity of the final selected regions in all known viral variants using tools like BLAST.
Detailed Protocol for Conservation Analysis
Method 1: Weighted, Scale-Agnostic Conservation Scoring This protocol is adapted from a method designed for low-variability genomes like SARS-CoV-2 [23].
- Objective: To identify contiguous regions of nucleic acid conservation that are independent of amino acid conservation.
- Input Data: A multiple sequence alignment (MSA) of a large number of viral genomes (e.g., thousands to millions).
- Software/Tools: Custom scripts (Python/R) for statistical analysis.
Procedure:
- Calculate Normalized Distance per Codon:
- For each codon position in a gene, compute a score based on the sum of Hamming distances (nucleotide differences) for every possible pair of sequences in the alignment.
- Normalize this score by dividing it by the expected score derived from the genome's overall codon usage, assuming the same amino acids were encoded. This controls for conservation driven purely by protein sequence constraints.
Apply Locus Weighting:
- Different codons provide different amounts of information about nucleic acid conservation beyond amino acid needs. For example, a locus where a single codon encodes a mandatory methionine provides zero information.
- Assign a weight to each locus proportional to the information it provides. This step is crucial in low-variability datasets to avoid artifacts.
Rank Data to Handle Skewness:
- In emerging viruses, most loci are highly conserved, making the few variable loci into strong outliers that can skew analysis.
- Transform the weighted conservation scores into ranks to move from a parametric to a non-parametric framework, improving robustness.
Identify Significant Regions:
- Use a scale-agnostic algorithm (e.g., based on finding the steepest descent in a random walk of the Z-statistics of the ranked data) to identify the most significantly conserved contiguous regions without imposing a fixed window size.
- Implement an iterative process to account for "interfering signals" – if one highly conserved region masks a second, the analysis is re-run with the first region removed to test for the significance of the second.
Table 1: Key Advantages of the Advanced Conservation Scoring Method
| Feature | Traditional Sliding Window | Advanced Method |
|---|---|---|
| Length Scale | Requires pre-defined, fixed window size | Scale-agnostic; finds regions of unexpected lengths |
| Information Use | Treats all loci equally | Weights loci by their informational content |
| Data Distribution | Assumes normal distribution | Uses ranking to handle skewed data from low-variability genomes |
| Multiple Signals | Can be masked by the strongest signal | Includes interference removal to find secondary conserved regions |
Method 2: Global Mutation Frequency Analysis This method provides a broader, gene-level overview of conservation.
- Objective: To determine the mutation frequency across all genes in a viral proteome.
- Input Data: A very large dataset of viral genome sequences (e.g., >10 million sequences from GISAID) [25].
- Software/Tools: Python packages for bioinformatics (e.g., NumPy, Pandas), alignment tools (e.g., EV couplings).
Procedure:
- Sequence Retrieval and Alignment: Download a comprehensive set of high-quality, full-length genome sequences. Align them to a reference genome.
- Variant Calling: Compare each sequence to the reference, identifying all amino acid-changing mutations.
- Calculate Mutation Frequency: For each gene or protein domain, calculate the frequency of mutations. A region is considered conserved if it shows no intergenic amino acid-changing replacements across the entire dataset.
- Identify Hotspots and Coldspots: Determine genomic regions with the highest (hotspots) and lowest (coldspots) mutation frequencies.
Table 2: Exemplary Mutation Frequency Data from a Global SARS-CoV-2 Analysis [25]
| Protein | Mutation Frequency Observation | Conservation Status |
|---|---|---|
| nsp11, nsp7, nsp10 | No mutations observed in >90% of sequences | Highly Conserved |
| nsp12 (RdRp) | P323L mutation present in 99.3% of sequences | Hotspot (but core enzyme is conserved) |
| Spike (S) | D614G mutation in 97.6% of sequences; highest mutation frequency in aa 508-635 | Variable Region |
| Nucleocapsid (N) | R203M mutation in 62.8% of sequences | Variable Region |
| Membrane (M) | Highest mutation frequency in aa 66-88 | Variable Region |
Experimental Validation and Toehold Switch Application
Selecting Targets for Toehold Switches
Once conserved regions are identified bioinformatically, they must be evaluated for suitability in toehold switch design.
- Avoid Mutational Hotspots: Intentionally design toehold binding regions to avoid structural protein genes susceptible to selective pressure from vaccination or infection [3].
- Ensure Specificity: Select regions that are conserved within the target virus but absent from closely related strains (e.g., specific to SARS-CoV-2 and not found in HCoV-OC43, HCoV-229E, etc.) [3].
- Avoid Secondary Structure: Analyze the target viral RNA region for local secondary structures (e.g., pseudoknots, UTRs) that might inhibit the toehold switch's trigger RNA from binding [3]. Use tools like NUPACK or ViennaRNA to predict secondary structure and normalized ensemble defect [14].
Protocol: Toehold Switch Sensor Design and Screening
This protocol outlines the development of a toehold switch for detecting a conserved viral RNA target [3] [14] [11].
- Objective: To create a functional toehold switch that produces a detectable signal upon binding to a conserved viral RNA trigger.
- Input: The conserved viral RNA sequence identified in Section 2.
Procedure:
- Design Toehold and Trigger:
- Design a 30-nucleotide trigger RNA sequence from the conserved viral target.
- Design the toehold switch RNA to include:
- A toehold (12 nt single-stranded region) complementary to the 5' end of the trigger.
- A stem (18-21 nt) that sequesters the Ribosome Binding Site (RBS) and start codon (AUG).
- A loop (11 nt) containing the RBS.
- Use design software (e.g., NUPACK, MeFit Toehold Designer) to model the switch's secondary structure and minimize normalized ensemble defect, ensuring efficient switching.
Clone into Expression Plasmid:
- Synthesize the toehold switch as part of a forward primer and clone it into a plasmid upstream of a reporter gene (e.g., mNeonGreen, lacZ) using restriction sites (e.g., XbaI, BamHI).
- The reporter gene should be chosen for high sensitivity (e.g., mNeonGreen is ~5x more intense than eGFP).
Test Specificity and Sensitivity In Vitro:
- Use a cell-free protein synthesis (CFPS) system to express the toehold switch.
- Add in vitro transcribed trigger RNA at a range of concentrations (e.g., from nM to fM) to test sensitivity.
- Measure reporter output (fluorescence for mNeonGreen/sfGFP, colorimetric change for LacZ/CPRG).
- Test against trigger RNAs from related viruses to confirm specificity and avoid cross-reactivity.
Incorporate Signal Amplification (For Diagnostics):
- To achieve clinically relevant sensitivity (femtomolar range), couple the toehold switch with an isothermal amplification step like NASBA or RT-LAMP, or a downstream enzymatic signal amplifier.
- Example (TEV Protease Amplification): Replace the fluorescent reporter with a TEV protease gene. When the toehold is activated, TEV protease is expressed and cleaves a quenched fluorescent substrate, with one protease molecule cleaving many substrates, leading to significant signal amplification [3].
The following workflow diagram illustrates the complete process from bioinformatic identification to functional validation of a toehold switch targeting a conserved viral region.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Toehold Switch Development
| Category | Item | Function/Application |
|---|---|---|
| Bioinformatics | Python (NumPy, Pandas), BLAST, NUPACK, ViennaRNA | Genome analysis, k-mer processing, sequence alignment, secondary structure prediction, and toehold switch design. |
| Cloning & Expression | High-fidelity DNA Polymerase (e.g., Phusion, Herculase II), Restriction Enzymes (e.g., NcoI, NotI, XbaI, BamHI), T4 DNA Ligase, pET28a or pUC19-based expression vectors | Amplification and assembly of DNA constructs; cloning toehold switches and reporter genes into plasmids. |
| In Vitro Transcription/Translation | T7 High Yield RNA Synthesis Kit, Cell-Free Protein Synthesis (CFPS) System (E. coli extract) | Producing trigger RNA and expressing the toehold switch/reporter system in a controlled, cell-free environment. |
| Reporter Systems | mNeonGreen/sfGFP: High-sensitivity fluorescent reporter.LacZ/CPRG: Colorimetric reporter (yellow to purple).TEV Protease + Quenched Substrate: Signal amplification system. | Providing a measurable output (fluorescence or color change) upon toehold switch activation. |
| Signal Amplification | NASBA or RT-LAMP Reagents, TEV Protease Cleavage Assay | Pre-amplification of target viral RNA or post-translational signal enhancement to achieve clinical-grade detection sensitivity. |
Targeting conserved, low-mutation regions is a foundational strategy for developing robust tools against evolving viruses. The integrated approach outlined here—combining advanced bioinformatic analyses of large-scale genomic datasets with rational toehold switch design and sensitive experimental validation—provides a reliable path forward. By focusing on regions of the viral genome that are under strong evolutionary constraint, researchers can create diagnostic sensors and therapeutic platforms with prolonged efficacy, ensuring they remain functional across diverse viral variants and contributing to more effective pandemic preparedness.
The programmability of RNA sensors, primarily through Watson-Crick base pairing, has established them as powerful tools for diagnostic and synthetic biology applications [26]. These systems function by undergoing a specific conformational change upon recognizing a trigger RNA, which then modulates the output of a reporter gene. The core challenge in designing these systems lies in achieving two often competing goals: high specificity for the intended target and robust performance that is resilient to interference from misfolded secondary structures. This document outlines validated strategies and detailed protocols for designing sensor and trigger RNAs, with a particular focus on the context of viral RNA detection using toehold switch technologies.
The design of these RNA components is critically influenced by the dynamics of RNA secondary structure. RNA molecules do not fold into a single structure but rather exist as an ensemble of conformations across a free energy landscape [27]. Successful sensor design must therefore navigate this landscape, strategically positioning the functional sensor within a low-energy minimum that is stable yet capable of undergoing a triggered conformational transition. This process is often co-transcriptional, meaning the RNA begins to fold while it is still being synthesized, which can lead to kinetic traps in non-functional conformations if not properly managed through sequence design [27].
Core Strategies for Enhancing Specificity
Fundamental Design Rules for Trigger-Sensor Interfaces
The interface between the sensor and its trigger RNA is the primary determinant of specificity. Meticulous design of this region can minimize off-target binding and ensure activation only by the intended RNA.
- Length and Complementarity: The sensor should be designed for extensive complementarity to its target viral RNA. For instance, toehold sensors used in SARS-CoV-2 detection were designed to have a sensing region that binds to a complementary "trigger" RNA, leading to a structural rearrangement that activates translation [28]. The typical length for an effective sensing domain can range from 51 to 75 nucleotides, with longer sequences sometimes offering better performance [29].
- Mechanism of Strand Displacement: Many advanced sensors, such as the Intelligent guide RNA (IngRNA) platform, utilize dual toehold switches. In this system, the binding of a trigger RNA to a first toehold site initiates a strand displacement cascade that releases a sequestered CrRNA, enabling it to function [7]. This multi-step mechanism provides an additional layer of specificity.
- Leveraging Ribosome Occupancy for Specificity: When designing sensors to detect endogenous cellular transcripts, the subcellular context and ribosome occupancy of the target mRNA are critical. Evidence suggests that ADAR-based editing sensors function more effectively when targeting sequences in the 3' untranslated region (3' UTR) or the coding sequence of a secreted protein, where ribosome occupancy is lower, as opposed to the coding sequence of a highly translated cytoplasmic protein. This can lead to activation ratios up to 45-fold higher [29].
Managing Secondary Structure and Energy Landscapes
The propensity of RNA to form stable secondary structures is a major challenge that can hinder the trigger-sensor interaction.
- Co-transcriptional Folding Considerations: Since RNA folds as it is transcribed, the order of sequence elements and the potential for intermediate structures must be considered. Transcription pausing, which can be influenced by RNA polymerase interactions, may be exploited to allow proper folding of functional domains [27].
- Navigating Kinetic Traps: RNA molecules can become trapped in local energy minima that are not the global minimum. These kinetic traps represent misfolded, non-functional states. Design strategies should aim to create an energy landscape where the functional state is both low in energy and accessible, with minimal energy barriers separating it from the non-functional state. The incorporation of toehold sequences is a key strategy here, as they provide an initial, weak binding site that facilitates the subsequent strand displacement reaction, effectively lowering the energy barrier for the conformational switch [7].
- Exploiting Conformational Dynamics: Two primary models describe how RNA sensors undergo structural changes: conformational selection and induced fit [27]. In conformational selection, the sensor spontaneously samples the active conformation, which is then stabilized by trigger binding. In the induced fit model, trigger binding itself induces the conformational change. Understanding these models helps in designing the sensor's aptamer or recognition domain; for example, ensuring that the trigger-binding site is accessible in a short-lived intermediate state can be a successful strategy, as seen in fluoride-sensing riboswitches [27].
Table 1: Computational Tools for RNA Design and Analysis
| Tool Name | Primary Function | Application in Sensor Design |
|---|---|---|
| NUPACK | Analysis of RNA secondary structure & strand interaction | Modeling the formation of complexes between sensor, trigger, and reporter RNAs; predicting secondary structures [28]. |
| PINTS | Identification of active promoters & enhancers from nascent transcript data | While focused on enhancer RNAs, its underlying principles highlight the importance of detecting unstable RNAs, analogous to some sensor triggers [30]. |
| RNA-MaP | High-throughput characterization of RNA-protein/dye binding | Massively parallel experimental measurement of sensor performance (e.g., Kd) across thousands of designs [31]. |
| Nucleologic | Automated design of nucleic acid sensors | Generating compact RNA (or DNA) sensors that compute complex functions of multiple inputs, as demonstrated for a tuberculosis diagnostic score [31]. |
Experimental Protocols for Sensor Validation
Protocol: In Vitro Characterization of RNA Sensor Function
This protocol details a method for testing sensor performance in a controlled cell-free environment, which is a critical first step before moving to cellular assays.
In Vitro Transcription of Sensor and Trigger RNAs:
- Design DNA templates for the sensor RNA and the target trigger RNA, each flanked by a T7 promoter sequence.
- Transcribe the RNAs in vitro using a T7 High Yield RNA Synthesis Kit according to the manufacturer's protocol.
- Treat the resulting RNA samples with DNase I for 30 minutes to remove any contaminating DNA template.
- Purify the transcribed RNAs using a standard method, such as phenol-chloroform extraction (e.g., TRIzol reagent) or a silica-membrane-based kit [7].
Cell-Free Translation Assay:
- Prepare a reaction mixture containing a cell-free translation system (e.g., E. coli S30 extract, wheat germ extract, or a commercial kit like the PURExpress system).
- To the reaction, add the purified sensor RNA and a range of concentrations of the purified trigger RNA (e.g., 0 nM, 10 nM, 100 nM, 200 nM).
- Incubate the reaction at a defined temperature (e.g., 37°C) for 1-2 hours to allow for translation.
- Measure the output signal. For a colorimetric output like LacZ, add a chromogenic substrate (e.g., ONPG) and measure the absorbance. For a luminescent output like nano-lantern, measure the luminescence directly [28].
Data Analysis:
- Plot the output signal (e.g., absorbance or luminescence units) against the concentration of the trigger RNA.
- Calculate the dynamic range of the sensor, typically defined as the fold-change in signal between the absence (0 nM) and presence (e.g., 200 nM) of the trigger RNA. The PHANTOM assay for SARS-CoV-2, for instance, achieved a sensitivity of 100 copies of viral RNA using a similar methodology [28].
Protocol: Validating Sensor Specificity via In Vitro Cleavage Assay
This protocol is particularly relevant for validating CRISPR-based guide RNA sensors, such as the IngRNA platform [7].
Protein and RNA Purification:
- Purify the relevant Cas protein (e.g., Cas9). This can be achieved by expressing a His-tagged protein in E. coli (e.g., strain BL21(DE3) pLysS) and purifying it using Ni-NTA affinity chromatography followed by cation-exchange chromatography [7].
- Transcribe and purify the guide RNA (e.g., IngRNA) and the trigger RNA in vitro, as described in Protocol 3.1.
- Prepare the target double-stranded DNA (dsDNA) by performing a PCR amplification of the target locus and purifying the product.
Cleavage Reaction:
- In a suitable reaction buffer (e.g., CutSmart Buffer), incubate 150 nM of the target dsDNA with 100 nM of the gRNA and 200 nM of the Cas9 protein.
- To test specificity, include reactions with and without the specific trigger RNA.
- Incubate the reaction at 37°C for 1-2 hours.
Analysis of Cleavage Efficiency:
- Stop the reactions with DNA loading dye.
- Analyze the products by non-denaturing agarose gel electrophoresis.
- Visualize the DNA bands under UV light. Efficient cleavage of the target DNA should be observed only in the presence of both the correct trigger RNA and the functional gRNA, confirming that the sensor is specifically activated by its intended trigger [7].
The Scientist's Toolkit: Essential Reagents and Solutions
Table 2: Key Research Reagent Solutions for RNA Sensor Development
| Reagent / Solution | Function | Example Use Case |
|---|---|---|
| T7 High Yield RNA Synthesis Kit | In vitro transcription of sensor, trigger, and reporter RNAs. | Generating high-quality, template-free RNA for functional assays [7]. |
| Cell-Free Translation System | In vitro protein synthesis to test sensor activity. | Validating that sensor activation leads to reporter protein (e.g., LacZ, luciferase) production [28]. |
| Chromogenic/Luminescent Substrates | Detection of reporter protein output. | ONPG for LacZ (colorimetric); luciferin for luciferase (luminescent) [28]. |
| Ni-NTA Agarose | Affinity purification of His-tagged proteins. | Purifying recombinant Cas proteins for in vitro cleavage assays [7]. |
| Isothermal Amplification Reagents | Amplification of low-abundance RNA targets. | NASBA enzymes for amplifying viral RNA to detectable levels in diagnostics [28]. |
Signaling Pathways and Workflow Visualizations
Isothermal amplification techniques have emerged as powerful tools for molecular diagnostics, offering rapid, sensitive, and specific detection of viral RNAs without the need for thermal cycling equipment. These characteristics make them particularly suitable for integration with novel biosensors, such as toehold switches, in point-of-care testing (POCT) platforms. Among these techniques, Nucleic Acid Sequence-Based Amplification (NASBA) and Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) have demonstrated exceptional utility for viral RNA detection due to their high sensitivity, specificity, and compatibility with colorimetric and fluorescent detection methods. This application note provides a detailed comparison of these two established isothermal amplification methods and presents optimized protocols for their implementation in viral detection assays, with particular emphasis on their coupling with toehold switch technology for the development of next-generation diagnostic platforms.
Performance Comparison: NASBA vs. RT-LAMP
The selection of an appropriate amplification strategy is critical for assay development. The table below summarizes key performance characteristics of NASBA and RT-LAMP based on published studies for viral RNA detection.
Table 1: Performance comparison of NASBA and RT-LAMP for viral RNA detection
| Parameter | NASBA | RT-LAMP |
|---|---|---|
| Optimal Temperature | 40-55°C [32] | 65°C [32] |
| Typical Amplification Time | 30-120 min [33] [34] | <60 min [35] |
| Reported Sensitivity | 0.5-2 copies/μL [33] [34] | 0.1 PFU [35] |
| Key Enzymes | AMV Reverse Transcriptase, RNase H, T7 RNA Polymerase [36] [33] | Bst DNA Polymerase, Reverse Transcriptase [35] |
| Primer Design Complexity | Two primers (one with T7 promoter) [37] | Four to six primers recognizing 6-8 regions [35] |
| Amplification Product | Single-stranded RNA [37] | Long, branched DNA with stem-loop structures [32] |
| Compatibility with Toehold Switches | Directly produces RNA for switch activation | Requires additional transcription step or specialized primer design |
Experimental Protocols
NASBA Protocol for Viral RNA Detection
The following protocol is adapted from established NASBA procedures with enhancements for toehold switch integration [36] [33] [38].
Reagent Preparation
- NASBA Buffer (5X concentrate): 200 mM Tris-HCl (pH 8.5), 60 mM MgCl₂, 250 mM KCl, 5 mM dNTPs, 10 mM NTPs [36]
- Primer Mix: 0.2 μM of each primer in nuclease-free water [36]
- Forward Primer: Contains target-binding sequence (e.g., norovirus GII ORF1-ORF2 junction region) and T7 promoter sequence at 5' end
- Reverse Primer: Contains target-binding sequence and optional G-quadruplex reverse complementary sequence for signal generation [33]
- Enzyme Mixture: 30% DMSO, 10 mM DTT, AMV Reverse Transcriptase (5 U/μL), RNase H (0.1 U/μL), T7 RNA Polymerase (40 U/μL) [36] [38]
- Enhancement Additive: T4 gene 32 protein (gp32) at 1 μg/reaction to eliminate thermal denaturation step [38]
Procedure
Reaction Assembly:
- Combine 5 μL of extracted RNA template with 5 μL of primer mix
- Add 10 μL of NASBA buffer (2X final concentration)
- Include 1 μL gp32 (1 μg/μL) for single-step protocol [38]
Amplification:
- Incubate at 41°C for 30-90 minutes
- For real-time monitoring, include molecular beacons or G4-ThT biosensor (4-6 μM ThT) in the reaction mix [33]
Detection:
- For toehold switch coupling, use 2-5 μL of NASBA product without purification
- Incubate with toehold switch reporter system at 37°C for 20 minutes
- Measure fluorescence or colorimetric signal
Critical Steps and Troubleshooting
- Primer Design: Ensure T7 promoter sequence is included in the forward primer; verify secondary structure formation
- Enzyme Handling: T7 RNA polymerase is thermolabile; maintain enzymes on ice until reaction initiation
- Magnesium Optimization: Adjust MgCl₂ concentration (8-12 mM final) if amplification efficiency is low
- Specificity Verification: Include no-template controls and heterologous viral RNA controls to confirm specificity
RT-LAMP Protocol for Viral RNA Detection
This protocol provides a standardized RT-LAMP procedure optimized for sensitivity and compatibility with downstream toehold switch sensors [35] [39].
Reagent Preparation
- Reaction Buffer (2X): 40 mM Tris-HCl (pH 8.8), 20 mM (NH₄)₂SO₄, 20 mM KCl, 8 mM MgSO₄, 0.2% Tween 20, 1.6 M betaine [35]
- Primer Set: Designed to recognize 6-8 distinct regions of target viral RNA using specialized software (e.g., PrimerExplorer V4) [40] [32]
- F3 (Forward outer primer): 18-22 nt
- B3 (Backward outer primer): 18-22 nt
- FIP (Forward inner primer): 40-45 nt containing F1C and F2 sequences
- BIP (Backward inner primer): 40-45 nt containing B1C and B2 sequences
- LF (Loop forward primer): 18-22 nt (optional, increases speed)
- LB (Loop backward primer): 18-22 nt (optional, increases speed)
- Enzyme Mixture: Bst DNA Polymerase (8 U/μL) and reverse transcriptase (e.g., AMV RT, 0.1 U/μL) [35]
Procedure
Reaction Setup:
- Combine 12.5 μL of 2X reaction buffer with 1-2 μL of primer mix (final: 1.6 μM FIP/BIP, 0.2 μM F3/B3, 0.4 μM LF/LB)
- Add 2 μL of target RNA and nuclease-free water to 22 μL total volume
- Heat mixture at 65°C for 1 minute to denature secondary structures
Amplification:
- Add 2 μL of enzyme mixture to reach 25 μL final volume
- Incubate at 63°C for 30-60 minutes
- Terminate reaction by heating at 80°C for 5 minutes
Toehold Switch Integration:
- For direct detection: Include toehold switch and reporter in RT-LAMP reaction
- For separate detection: Dilute RT-LAMP product 1:10 and add to toehold switch reaction
- Incubate at 37°C for 15-30 minutes before signal measurement [39]
Critical Steps and Troubleshooting
- Primer Design: Use dedicated software and verify specificity against target sequence; avoid primer dimerization
- Temperature Calibration: Verify incubator temperature accuracy (±0.5°C) as Bst polymerase is temperature-sensitive
- Magnesium Concentration: Optimize MgSO₄ concentration (4-8 mM) if precipitation or poor amplification occurs
- Inhibition Management: For complex samples, use 1-2% BSA or 0.1-0.5 M trehalose to overcome inhibition
Research Reagent Solutions
The table below outlines essential reagents and their functions for implementing NASBA and RT-LAMP assays in research settings.
Table 2: Essential research reagents for NASBA and RT-LAMP assays
| Reagent | Function | Application Notes |
|---|---|---|
| Bst DNA Polymerase, Large Fragment | Strand-displacing DNA polymerase for LAMP amplification | Maintains activity at 65°C; high processivity enables efficient amplification [32] |
| AMV Reverse Transcriptase | RNA-directed DNA polymerase for cDNA synthesis | Used in both NASBA and RT-LAMP; thermostable variants preferred for RT-LAMP [33] [32] |
| T7 RNA Polymerase | DNA-directed RNA polymerase for NASBA amplification | Generates multiple RNA copies from DNA template; thermolabile (requires <50°C) [36] [32] |
| RNase H | Ribonuclease that degrades RNA in RNA-DNA hybrids | Essential for NASBA cycle; enables recycling of template [36] [33] |
| T4 Gene 32 Protein (gp32) | Single-stranded binding protein | Enhances NASBA efficiency; enables single-pot reaction by eliminating thermal denaturation step [38] |
| Betaine | Chemical additive | Reduces secondary structure formation in DNA; essential for LAMP efficiency [35] |
| Molecular Beacons | Fluorescent hybridization probes | Enable real-time monitoring of NASBA amplification; highly specific due to stem-loop structure [38] |
| Thioflavin T (ThT) | G-quadruplex fluorescent biosensor | Binds G4 structures in NASBA products; >1700x fluorescence enhancement enables sensitive detection [33] |
Workflow Integration with Toehold Switches
The integration of isothermal amplification with toehold switch technology creates a powerful biosensing platform for viral RNA detection. The diagrams below illustrate the conceptual workflow for coupling each amplification method with toehold switches.
NASBA-Toehold Switch Workflow
NASBA to Toehold Switch Detection Workflow
RT-LAMP-Toehold Switch Workflow
RT-LAMP to Toehold Switch Detection Workflow
NASBA and RT-LAMP represent two powerful isothermal amplification technologies with complementary strengths for viral RNA detection. NASBA offers direct RNA amplification, making it inherently compatible with RNA-based toehold switches without requiring additional transcription steps. Its high sensitivity (0.5-2 copies/μL) and single-stranded RNA products facilitate seamless integration with toehold switch technology. Recent enhancements, such as the incorporation of T4 gene 32 protein, have simplified the NASBA workflow to a single-pot reaction, improving its suitability for point-of-care applications [38].
RT-LAMP provides rapid amplification (<60 minutes) with exceptional sensitivity (0.1 PFU), but requires specialized primer design and may need additional optimization for toehold switch integration [35]. The higher operating temperature (65°C) of RT-LAMP enhances specificity but may present challenges for some toehold switch configurations.
For researchers developing toehold switch-based viral detection platforms, NASBA offers a more direct integration pathway due to its native production of RNA amplicons. The recent development of NESBA (nicking and extension chain reaction system-based amplification) further enhances NASBA's capabilities, achieving detection limits of 0.5 copies/μL within 30 minutes while maintaining 100% clinical sensitivity and specificity for SARS-CoV-2 [34]. Both platforms provide robust, sensitive alternatives to PCR-based methods and continue to evolve through enzyme engineering and protocol optimization, offering powerful tools for the next generation of molecular diagnostics.
Reporter systems are indispensable tools in molecular biology, enabling the visualization and quantification of biological events such as gene expression and pathogen detection. This Application Note details the use of colorimetric (LacZ/CPRG) and fluorescent (sfGFP, mNeonGreen) reporters, with a specific focus on their integration into toehold switch platforms for sensitive viral RNA detection. The protocols and data presented herein provide a framework for researchers to implement these robust systems in their laboratories.
Research Reagent Solutions
The following table catalogues the essential reagents and their functions for implementing toehold switch-based reporter assays.
Table 1: Key Research Reagents for Toehold Switch Reporter Assays
| Item | Function/Description |
|---|---|
| Toehold Switch RNA | Synthetic riboregulator; hairpin structure blocks translation initiation; unfolds upon binding specific viral RNA trigger [16]. |
| LacZ Reporter Gene | Encodes β-galactosidase enzyme; enables colorimetric readout with substrates like CPRG [16]. |
| CPRG Substrate | Chlorophenol-red-β-D-galactopyranoside; yellow substrate cleaved by β-galactosidase to produce a red dye for visible colorimetric detection [16]. |
| mNeonGreen | Bright, fast-maturing fluorescent protein; ideal for real-time, quantitative reporting with high spatial/temporal resolution [41]. |
| sfGFP Variants | Engineered superfolder GFP; superior folding and solubility; selected variants offer reliable functionality in thermophilic bacteria [42]. |
| BacMam Delivery System | Modified baculovirus for efficient sensor delivery and expression in a wide variety of mammalian cell types [43]. |
| sGC (Soluble Guanylate Cyclase) | Used as a positive control; produces cGMP in response to Nitric Oxide, validating sensor function [43]. |
| Sodium Butyrate/Valproic Acid | HDAC inhibitors; enhance and maintain BacMam-driven expression in transduced cells [43]. |
Quantitative Comparison of Reporter Systems
The choice between colorimetric and fluorescent reporters depends on the application's specific requirements for sensitivity, quantitation, and throughput.
Table 2: Performance Characteristics of Reporter Systems
| Reporter System | Key Features | Maturation Time | Dynamic Range / Sensitivity | Primary Applications |
|---|---|---|---|---|
| LacZ/CPRG (Colorimetric) | Output: Yellow to red color change. Equipment: Simple plate reader or visual inspection. | Enzyme-dependent; typically fast. | Demonstrated in sensitive paper-based viral RNA sensors [16]. | Portable diagnostics, endpoint assays in resource-limited settings [16]. |
| sfGFP (Fluorescent) | Excitation/Emission: ~485-510 nm. Brightness: High. | Fast-folding variant of GFP [42]. | 885-fold improved MFI in thermophilic bacteria after engineering (variant: sfGFP(N39D/A179A)) [42]. | Real-time monitoring in thermophilic bacteria; flow cytometry; promoter activity studies [42]. |
| mNeonGreen (Fluorescent) | Excitation/Emission: ~506/517 nm. Brightness: High. | ~7 minutes [41]. | Higher detection sensitivity vs. MS2-MCP system; enables tracking of weak expression patterns [41]. | Live-cell imaging; quantitative tracking of rapid transcriptional dynamics in development [41] [43]. |
Application Notes & Experimental Protocols
Protocol: Paper-Based Toehold Switch Assay for Viral RNA (Colorimetric Readout)
This protocol is adapted from a study detecting Zika and novel coronaviruses using a toehold switch and LacZ/CPRG output [16].
Workflow Overview:
Procedure:
- Sensor Immobilization: Pre-immobilize the toehold switch RNA and necessary cell-free protein synthesis machinery (ribosomes, tRNAs, etc.) onto a paper matrix.
- Sample Application: Apply the processed sample, potentially containing the viral RNA trigger, to the paper strip.
- Incubation: Allow the reaction to proceed at room temperature (e.g., 30-60 minutes). If the target viral RNA is present, it will bind the toehold switch, unlocking the ribosome binding site and initiating translation of the
LacZreporter gene. - Color Development: Add the CPRG substrate. The synthesized β-galactosidase enzyme will cleave CPRG, converting the solution from yellow to red.
- Detection: Visual assessment of the color change or quantification using a simple scanner or portable spectrophotometer.
Protocol: Live-Cell Fluorescent Reporter Assay with mNeonGreen
This protocol for measuring cell signaling dynamics in live cells can be adapted for viral sensor readouts using the bright mNeonGreen protein [41] [43].
Workflow Overview:
Procedure:
- Cell Preparation (Day 1):
- Harvest adherent cells (e.g., HEK293T) using a standard trypsinization protocol. Resuspend the cell pellet in complete culture media and determine cell concentration.
- Prepare a cell suspension at 500,000 cells/mL in complete media. Scale the volume according to the number of wells (e.g., 100 μL/well for a 96-well plate).
Viral Transduction (Day 1):
- For each well, prepare a transduction mix containing:
- 25 μL mNeonGreen Sensor BacMam stock.
- 0.6 μL of 500 mM Sodium Butyrate stock (final conc. ~2 mM).
- 24.4 μL complete culture media.
- Gently mix 100 μL of cell suspension (Tube A) with 50 μL of transduction mix (Tube B) and seed the entire 150 μL into a 96-well plate.
- Cover the plate with foil and incubate for 30 minutes at room temperature.
- Transfer the plate to a 37°C CO₂ incubator and culture for 20-24 hours.
- For each well, prepare a transduction mix containing:
Fluorescence Measurement (Day 2):
- Gently replace the culture media with Dulbecco's Phosphate Buffered Saline (DPBS) containing calcium and magnesium.
- Allow the cells to rest in DPBS for 25-40 minutes at room temperature before reading to stabilize the baseline.
- Measure fluorescence using a plate reader or microscope configured with standard GFP filter sets (Excitation ~506 nm, Emission ~517 nm). For cGMP sensors, a decrease in mNeonGreen fluorescence indicates rising cGMP levels [43].
Notes on sfGFP for Use in Thermophilic Systems
When working with thermophilic bacteria (e.g., Parageobacillus thermoglucosidasius), standard FPs misfold at high temperatures. Use an engineered sfGFP variant like sfGFP(N39D/A179A), which provides an 885-fold enhanced mean fluorescence intensity at 60°C [42]. Ensure the expression vector is optimized for the host, using validated thermophilic promoters and codon-optimization if necessary.
The detection of viral RNA is a critical capability for public health, diagnostic medicine, and pandemic preparedness. Toehold switches represent a breakthrough in synthetic biology, serving as de-novo-designed riboregulators that activate gene expression in response to specific RNA sequences [1]. When combined with paper-based, cell-free biosensor systems, these components create a powerful platform for diagnostic applications outside conventional laboratory settings [44] [45]. This integration enables the development of portable, low-cost detection systems that can be deployed for viral RNA detection in resource-limited environments. These systems bypass the need for living, genetically modified organisms, thereby overcoming significant limitations in shelf life, usability, and biosafety [44]. The resulting platform offers researchers and public health professionals a rapid, field-deployable tool for identifying viral pathogens, with particular relevance for emerging respiratory threats [46] [47].
System Components and Integration
The integrated platform comprises three essential elements: the paper-based biosensor, the toehold switch mechanism, and the portable detection device. This configuration creates a complete sample-to-answer system that requires minimal technical expertise to operate.
Table 1: Core Components of the Integrated Detection Platform
| Component Category | Specific Elements | Function in Detection System |
|---|---|---|
| Paper-Based Biosensor | Freeze-dried cell-free protein synthesis (CFPS) system [44] | Provides stable, room-temperature storage of biochemical reagents; rehydrates with aqueous sample to initiate reaction |
| Molecular Recognition | Toehold switch RNA [1] | Acts as biosensor; binds specifically to target viral RNA sequence and triggers translation of reporter protein |
| Signal Generation | Superfolder green fluorescent protein (sfGFP) [44] | Serves as reporter molecule; produces measurable fluorescence signal upon toehold switch activation |
| Detection Hardware | Smartphone with custom filters [44] | Provides portable, accessible platform for fluorescence detection and signal quantification |
| Sample Interface | Paper matrix [44] | Acts as support for freeze-dried reagents; creates uniform reaction environment upon sample addition |
Molecular Mechanism of Toehold Switches
Toehold switches are synthetic RNA regulators designed to detect specific RNA sequences with high specificity and minimal cross-reactivity. These switches employ a strand displacement mechanism that differs fundamentally from natural riboregulators [1]. In the absence of the target viral RNA, the toehold switch maintains a hairpin secondary structure that sequesters the ribosome binding site (RBS) and start codon, effectively preventing translation initiation. When the target viral RNA is present, it binds to a complementary "toehold" region—a single-stranded linear sequence—initiating a thermodynamically favorable strand displacement that unfolds the hairpin and exposes the RBS. This structural rearrangement enables ribosome access and initiates translation of the reporter protein (sfGFP), generating a measurable signal [1]. This mechanism provides exceptional dynamic range (often exceeding 400-fold) and high orthogonality, allowing simultaneous detection of multiple viral targets without significant crosstalk [1].
Diagram 1: Workflow of the integrated paper-based biosensor system for viral RNA detection, showing both positive (signal generation) and negative (no signal) outcomes.
Materials and Reagents
Research Reagent Solutions
Table 2: Essential Research Reagents for Toehold Switch-Based Detection
| Reagent/Category | Specific Examples | Function in Experimental System |
|---|---|---|
| Cell-Free Protein Synthesis System | E. coli extract [44] | Provides transcriptional and translational machinery for protein synthesis without intact cells |
| Toehold Switch Plasmids | Custom-designed constructs [1] | Encode toehold switch sequence and reporter gene; can be forward-engineered for specific viral targets |
| Reporter Protein | Superfolder GFP (sfGFP) [44] | Fluorescent reporter with rapid folding and stability under diverse conditions |
| Paper Matrix | Chromatography or filter paper [44] | Serves as solid support for freeze-dried reaction components; enables capillary flow |
| RNA Extraction Reagents | Guanidine-based solution [47] | Inactivates and stabilizes viral nucleic acids from clinical or environmental samples |
| Positive Control Templates | Synthetic viral RNA targets [1] | Validate system functionality and establish detection limits for target viruses |
Experimental Protocol
Biosensor Preparation and Assembly
Day 1: Toehold Switch Design and Preparation
- Design toehold switch sequences specific to target viral RNA using established algorithms [1]. The trigger binding domain should be perfectly complementary to a unique ~30 nt region of the target viral genome.
- Clone designed switches into appropriate expression vectors containing a T7 promoter and the sfGFP reporter gene [44] [1]. Verify constructs through sequencing.
- Prepare cell-free reaction mixture containing E. coli extract, energy sources (ATP, GTP), amino acids, RNA polymerase, and necessary salts [44].
- Combine cell-free mixture with plasmid DNA encoding the toehold switch and dispense 10-15 µL aliquots onto paper strips (approximately 0.5 cm²).
- Freeze-dry prepared paper strips overnight to remove all moisture while preserving biochemical activity [44].
Day 2: Sample Processing and RNA Extraction
- Collect environmental samples using synthetic-tip swabs moistened with DNase/RNase-free water [47]. Swab approximately 100 cm² of surface area using horizontal and vertical motions.
- Place swabs in tubes containing 500 µL guanidine-based viral transport medium to inactivate and stabilize viral RNA [47].
- Extract RNA using commercial viral nucleic acid isolation kits [47]. Elute in 50 µL nuclease-free water.
- Add process controls such as synthetic RNA to monitor extraction efficiency and detect PCR inhibitors [47].
Detection Procedure and Data Analysis
Day 2: Assay Execution and Signal Detection
- Rehydrate paper sensors with 10 µL of extracted RNA sample. Include positive controls (synthetic target RNA) and negative controls (nuclease-free water).
- Incubate rehydrated sensors at 37°C for 60-90 minutes to allow for toehold switch activation and sfGFP production [44].
- Place reacted sensors in a dark chamber or box to minimize ambient light interference.
- Use smartphone with custom filter system (blue LED excitation filter ~485 nm, emission filter ~510 nm) to capture fluorescence images [44].
- Analyze images using mobile applications that quantify green fluorescence intensity relative to negative controls.
Interpretation and Quality Control
- Calculate signal-to-noise ratio by dividing fluorescence intensity of samples by intensity of negative controls.
- Establish positive threshold typically at 3-5 times the standard deviation above mean negative control values.
- Validate assay performance with known positive and negative samples to determine clinical sensitivity and specificity.
- Monitor process controls to identify potential inhibition or extraction failures [47].
Performance Characterization
Analytical Sensitivity and Specificity
Table 3: Performance Metrics of Toehold Switch-Based Detection Systems
| Performance Parameter | Reported Values | Experimental Conditions |
|---|---|---|
| Dynamic Range | >400-fold activation [1] | Comparison of fluorescence signal between presence and absence of target RNA |
| Detection Limit | 6 μg/L (Mercury model) [44] | Demonstrates high sensitivity of cell-free biosensor platform |
| Orthogonality | 26 simultaneous targets with <12% crosstalk [1] | Capability for multiplexed detection without signal interference |
| Time to Result | 60-90 minutes [44] | From sample application to measurable signal output |
| Storage Stability | Several weeks (freeze-dried) [44] | Room temperature storage on paper matrix |
| Environmental Detection | 16% positivity rate on surfaces [47] | Detection of viral RNA on high-touch surfaces in university settings |
The platform demonstrates particular strength in multiplexing capability, enabling the simultaneous detection of multiple viral targets—a critical feature for comprehensive respiratory virus surveillance where pathogens like SARS-CoV-2, influenza, and RSV often co-circulate [47]. The orthogonal nature of toehold switches allows researchers to design panels for parallel detection without significant signal interference [1]. Furthermore, the system's compatibility with environmental sampling extends its utility beyond clinical diagnostics to public health surveillance, enabling monitoring of viral contamination on high-touch surfaces in community settings [47].
Diagram 2: Molecular mechanism of toehold switch activation by target viral RNA, showing the transition from inactive hairpin to open structure enabling reporter protein translation.
Applications in Viral RNA Detection
The integration of paper-based cell-free systems with portable readers creates a versatile platform with multiple applications in viral research and public health:
Respiratory Virus Surveillance: Simultaneous detection of SARS-CoV-2, influenza A/B, and RSV A/B from environmental samples [47]. The platform's multiplexing capability enables comprehensive monitoring of co-circulating respiratory pathogens in high-transmission settings like universities and healthcare facilities.
Outbreak Investigation: Rapid deployment for identifying viral hotspots through environmental monitoring of high-touch surfaces (doorknobs, desks, handles) [47]. The system provides an early warning mechanism for viral circulation before clinical cases escalate.
Therapeutic Development: Screening potential RNA-targeting compounds like mirafloxacin and "Compound 6" that disrupt viral frameshift elements or other structured RNA motifs [48]. The platform enables medium-throughput evaluation of antiviral candidates.
Field-Based Diagnostics: Deployment in resource-limited settings where traditional laboratory infrastructure is unavailable. The system's freeze-dried format and smartphone-based detection eliminate needs for cold chain and specialized equipment [44].
Viral Evolution Tracking: Adaptation to detect emerging variants through redesign of toehold switch trigger sequences. The programmability of the platform allows rapid response to viral mutations that evade existing detection methods [46].
Troubleshooting and Optimization
Common challenges in implementing the integrated platform and recommended solutions:
Low Signal Intensity: Optimize toehold switch design by adjusting the length and stability of the stem region. Increase the concentration of cell-free reaction components, particularly energy sources and RNA polymerase [44].
High Background Signal: Include additional negative control switches with scrambled sequences to assess non-specific activation. Optimize paper matrix composition to reduce non-specific binding [1].
Inconsistent Results Between Replicates: Standardize freeze-drying protocol to ensure uniform reagent distribution. Implement quality control measures for RNA extraction efficiency [47].
Limited Multiplexing Capacity: Design toehold switches with minimal sequence similarity to reduce crosstalk. Balance expression levels of different reporters to prevent competition for translational resources [1].
Short Shelf Life: Include stabilizers such as trehalose in the freeze-drying formulation. Package sensors with desiccant to maintain dryness during storage [44].
Toehold switches are synthetic riboregulators that provide a programmable and highly specific method for RNA detection, making them invaluable tools in molecular diagnostics and synthetic biology. These RNA-based biosensors operate through a conformational change: their hairpin structure, which sequesters the ribosome binding site (RBS) and start codon, unfolds upon binding to a specific trigger RNA, thereby initiating translation of a reporter gene [3] [49]. The primary advantage of toehold switches lies in their design flexibility, which allows them to be tailored to detect virtually any RNA sequence, from viral pathogens to endogenous genetic markers. This application note details successful implementations of toehold switch technology for detecting human viruses such as SARS-CoV-2 and Zika, as well as critical plant pathogens, providing researchers with validated protocols and frameworks for developing their own detection systems.
SARS-CoV-2 Detection with Downstream Signal Amplification
Experimental Approach and Design Rationale
A cell-free toehold switch-based biosensor was developed to detect SARS-CoV-2 RNA without requiring upstream RNA amplification. The design process focused on identifying optimal target regions within the viral genome. Researchers selected the nonstructural protein 2 (Nsp2) region of the SARS-CoV-2 genome (Wuhan strain, NCBI Reference Sequence NC_045512.2) due to its low mutation rate and conservation among SARS-CoV-2 variants, while being distinct from human coronaviruses that cause mild respiratory diseases (HCoV-OC43, HCoV-229E, and HCoV-NL6) [3]. This strategic selection ensured the biosensor would remain effective despite expected viral mutations and avoid cross-reactivity.
Toehold switches were designed with a 3-nucleotide GGG T7 promoter enhancer sequence, a 12-nucleotide toehold region, and an 18-nucleotide stem sequence complementary to a 30-nucleotide RNA trigger. This was followed by an 11-nucleotide loop containing the RBS and another 18 nucleotides complementary to the first stem segment, with a start codon positioned 6 nucleotides after the loop [3]. The designs were computationally screened to avoid regions with interfering secondary structure in the viral RNA binding region.
Key Findings and Performance Metrics
The optimized toehold switch (CSU 08) demonstrated detection sensitivity in the low picomolar range for target RNA when using the highly sensitive fluorescent reporter mNeonGreen, which provides approximately five times greater intensity than traditional eGFP [3]. To achieve clinically relevant sensitivity, researchers implemented a modular downstream amplification system where toehold switch activation controls expression of tobacco etch virus (TEV) protease. The TEV protease then cleaves a quenched fluorescent reporter, enabling significant signal amplification. This approach enhanced sensitivity to the low femtomolar range for target RNA detection, eliminating the need for upstream nucleic acid amplification [3].
Table 1: Performance Metrics for SARS-CoV-2 Toehold Switch Detection
| Component | Parameter | Performance |
|---|---|---|
| Toehold Switch CSU 08 | Detection Sensitivity (with mNeonGreen) | Low picomolar range |
| With TEV Protease Amplification | Detection Sensitivity | Low femtomolar range |
| Reporter Protein | mNeonGreen Intensity vs eGFP | ~5x more intense |
| Target Region | SARS-CoV-2 Nsp2 | Low mutation region |
Detailed Protocol
Step 1: Toehold Switch Design and Construction
- Design toehold switches using the STORM toehold switch generator software or manual iterative design based on the target SARS-CoV-2 sequence.
- Synthesize toehold switches as 117-nt forward primers using ultramer DNA oligonucleotides containing an XbaI site and a 19-nt region complementary to the mNeonGreen gene.
- Clone toehold switches into the pUC19-mNeonGreen expression plasmid downstream of a T7 phage promoter using XbaI and HindIII restriction sites [3].
Step 2: Cell-Free Protein Synthesis System Preparation
- Prepare E. coli-based cell-free protein synthesis (CFPS) system according to established protocols.
- Lyophilize CFPS reagents on paper substrates for improved stability and point-of-care application.
- For saliva samples, add murine RNase inhibitor (mRI) to the CFPS system to maintain reaction robustness [50].
Step 3: Detection Reaction Assembly
- Combine lyophilized CFPS reagents with toehold switch plasmid (10-20 nM final concentration).
- Add sample containing potential SARS-CoV-2 RNA trigger (15 μL saliva can be used directly).
- Incubate at 37°C for 30-90 minutes depending on required sensitivity.
- For amplified detection, use the TEV protease system where toehold switch activates TEV protease expression, which then cleaves a quenched fluorescent reporter [3].
Step 4: Signal Detection
- Monitor fluorescence using a plate reader (mNeonGreen: excitation 506 nm, emission 517 nm).
- For visual detection, use a bioluminescent reporter (NanoLuc) that produces visible light in presence of target RNA [50].
- The TEV protease amplification system provides greater fold change between control and sample [3].
Diagram 1: SARS-CoV-2 detection workflow using toehold switches with optional signal amplification.
Plant Pathogen Detection Using Toehold Switches
Turnip Mosaic Virus (TuMV) Detection in Pseudostellaria heterophylla
Plant pathogens cause significant agricultural losses worldwide, and rapid detection is crucial for effective disease management. A toehold switch-based system was developed to detect Turnip mosaic virus (TuMV) in Pseudostellaria heterophylla, a traditional Chinese medicinal herb [11]. Researchers designed toehold switches targeting conserved regions of the TuMV genome (P1, HC-pro, and P3 coding regions) with high specificity to the Potyvirus genus and no sequence similarity to the P. heterophylla genome.
The detection platform integrated nucleic acid sequence-based amplification (NASBA) for RNA preamplification, followed by toehold switch activation in a cell-free system. The selected sensor demonstrated detection of 1 pM TuMV RNA fragments within 40 minutes, with a detection limit of less than 10 fM when the reaction time was extended to 90 minutes [11]. The system showed high specificity with no cross-reactivity against cucumber mosaic virus, another prevalent viral pathogen in P. heterophylla.
Rosewood Species Identification
Toehold switches have also been applied to conservation efforts through the detection of endangered rosewood species (Dalbergia maritima). Researchers developed a streamlined pipeline for creating RNA-based sensors that distinguish this protected species from other timber sources [49]. Target sequences from the MatK, RbcL, and TrnL-UAA genes were used to design species-specific toehold switches.
The rosewood toehold switches were designed using the Chinese University of Hong Kong (CUHK) model, adopting the optimized Series B sensor architecture previously used for Zika virus detection [49]. Switches were assembled in low-copy plasmids upstream of sfGFP fused to an LVA degradation tag, with expression controlled by a T7 promoter. Functional sensors were identified through in vivo screening in E. coli BL21 Star(DE3) cells, which have reduced mRNA degradation due to a truncated RNaseE gene.
Table 2: Plant Pathogen Detection Using Toehold Switches
| Pathogen/Application | Detection Limit | Time | Amplification Method | Reporter |
|---|---|---|---|---|
| Turnip Mosaic Virus (TuMV) | 10 fM | 90 min | NASBA | LacZ/CPRG (colorimetric) |
| Rosewood (Dalbergia maritima) | Not specified | Not specified | None (direct detection) | sfGFP (fluorescence) |
| Cucumber Green Mottle Mosaic Virus | Designed | Not tested | RPA | CDO (colorimetric) |
| Barley Yellow Dwarf Virus | Designed | Not tested | Not specified | LacZ/mScarlet-I |
Detailed Protocol for Plant Pathogen Detection
Step 1: Sample Preparation from Plant Material
- Grind 100 mg of plant leaf tissue using a mechanical disruptor in 500 μL of lysate buffer.
- Centrifuge at 12,000 × g for 5 minutes to pellet debris.
- Use supernatant directly for amplification without nucleic acid purification [51].
Step 2: Nucleic Acid Amplification
- For NASBA: Combine 5 μL of plant lysate with 40 μL of NASBA reaction mixture containing primers targeting the pathogen sequence.
- Incubate at 41°C for 90 minutes for isothermal amplification [11].
- Alternatively, use Recombinase Polymerase Amplification (RPA) with body heat incubation (37°C) for 30 minutes [51].
Step 3: Toehold Switch Activation
- Combine 10 μL of amplification product with cell-free expression system containing toehold switch plasmid.
- Add substrate for colorimetric detection (CPRG for LacZ or catechol for CDO).
- Incubate at 37°C for 30-150 minutes depending on required sensitivity [11] [51].
Step 4: Result Interpretation
- Visual inspection for color change: yellow for CDO/catechol, purple for LacZ/CPRG.
- For quantitative results, measure absorbance at 570 nm (CPRG) or 425 nm (catechol) [11] [51].
- Compare to positive and negative controls to confirm specific detection.
Zika Virus Detection Platform
Experimental Design and Implementation
The Zika virus detection platform represents one of the earliest and most influential applications of toehold switches for viral diagnostics. This system employed toehold switches designed to recognize specific sequences within the Zika virus genome, coupled with upstream nucleic acid sequence-based amplification (NASBA) to achieve the necessary sensitivity for clinical detection [3].
The toehold switches followed the Series B sensor architecture, which features a stem-loop structure that sequesters the RBS and start codon, preventing translation in the absence of the trigger RNA. When the target Zika virus RNA is present, it binds to the toehold region and unwinds the hairpin through strand displacement, exposing the RBS and initiating translation of a reporter protein [49]. The system was implemented in a paper-based cell-free format that could be lyophilized for storage and distribution, making it suitable for field use in resource-limited settings.
Performance and Impact
The Zika virus detection system demonstrated a sensitivity of 3 fM for synthetic trigger RNA when combined with NASBA pre-amplification, a significant improvement over the 30 nM sensitivity of the toehold switch alone [3]. This level of sensitivity enabled detection of clinically relevant concentrations of viral RNA in patient samples.
The platform utilized a colorimetric output based on β-galactosidase (LacZ) expression, which hydrolyzes chlorophenol red-β-D-galactopyranoside (CPRG) from yellow to purple, allowing visual detection without instrumentation [3] [49]. This approach established the foundation for subsequent toehold switch-based diagnostics and demonstrated the potential for low-cost, point-of-care nucleic acid detection.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Toehold Switch Development
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Toehold Switch Design Tools | STORM Toehold Switch Generator, NUPACK, MeFit Toehold Designer | Computational design of optimal toehold switch sequences and prediction of RNA secondary structures |
| Cell-Free Expression Systems | E. coli lysate-based CFPS, PURExpress | In vitro transcription and translation without intact cells |
| Reporter Systems | mNeonGreen, sfGFP, LacZ/CPRG, NanoLuc, CDO/catechol | Signal generation through fluorescence, colorimetric change, or bioluminescence |
| Amplification Methods | NASBA, RT-LAMP, RPA | Pre-amplification of target RNA for enhanced sensitivity |
| Specialized Reagents | Murine RNase Inhibitor (mRI), TEV Protease system | Enhanced stability in complex samples (e.g., saliva), signal amplification |
| Vector Systems | pUC19-mNeonGreen, pSB3T5, pET expression vectors | Toehold switch and reporter gene expression |
Toehold switches represent a versatile and powerful platform for nucleic acid detection across diverse applications from clinical diagnostics to agricultural biotechnology and conservation biology. The case studies presented here demonstrate consistent performance characteristics: high specificity through programmable sequence recognition, sensitivity enhanced through various amplification strategies, and adaptability to different output modalities for point-of-use testing. As the field advances, integration of machine learning approaches like Toehold-VISTA for improved design and continued refinement of signal amplification strategies will further enhance the capabilities of these synthetic biology tools [12]. The protocols and reagents detailed in this application note provide researchers with a foundation for developing toehold switch-based detection systems for their specific targets of interest.
Diagram 2: Molecular mechanism of toehold switch activation and reporter gene expression.
Enhancing Performance: Strategies for Troubleshooting and Signal Amplification
Toehold switches represent a powerful class of de-novo-designed prokaryotic riboregulators that activate gene expression in response to cognate RNA triggers, demonstrating high orthogonality and an average dynamic range above 400-fold [1]. These programmable RNA switches have become a cornerstone in synthetic biology for detecting viral RNAs, yet their ultimate sensitivity in diagnostic applications often depends on the efficiency of the downstream reporter system [15] [13]. This application note explores the integration of Tobacco Etch Virus protease (TEVp) as a critical signal amplification component in toehold switch-based detection platforms.
The wild-type TEV protease, while highly sequence-specific, suffers from a slow catalytic rate ((k_{cat}) of 0.18 s⁻¹), fundamentally limiting its ability to rapidly process and amplify detection signals [52]. Recent protein engineering efforts have addressed this limitation through directed evolution, producing TEVp variants with significantly enhanced catalytic efficiency. When incorporated into synthetic genetic circuits, these engineered proteases enable substantial improvements in temporal resolution and signal-to-background ratios [52] [53]. This protocol details the implementation of evolved TEV protease systems for boosting detection sensitivity in viral RNA diagnostics employing toehold switch technology.
Technical Specifications: Evolved TEV Protease Variants
Directed evolution of TEV protease has yielded several variants with improved catalytic properties. The selection platform utilized a yeast-based system where protease activity was coupled to the release of a membrane-anchored transcription factor via a photo-inducible mechanism [52]. Through successive rounds of selection with decreasing light exposure times, researchers enriched faster TEVp variants with mutations surrounding the catalytic triad.
Table 1: Engineered TEV Protease Variants and Their Catalytic Improvements
| Variant Name | Mutations | Catalytic Efficiency | Key Applications | Signal-to-Background Improvement |
|---|---|---|---|---|
| uTEV1Δ | S153N | Significantly improved | FLARE, SPARK | 27-fold over wild-type |
| uTEV2Δ | T30A/S153N | Significantly improved | Calcium integration | Not specified |
| TEVp-C1 | Multiple distal mutations | Enhanced for non-canonical P1' residues | Traceless cleavage applications | Not specified |
| stTEVp | T17S, N68D, I77V, R203G, S219N | Improved solubility and activity | General protein purification | Not specified |
The S153N mutation (uTEV1Δ) was particularly noteworthy, as when incorporated into the calcium integrator FLARE, it improved the signal-to-background ratio by 27-fold and enabled recording of neuronal activity with 60-second temporal resolution [52]. This substantial improvement in signal amplification capacity makes uTEV1Δ particularly valuable for enhancing the sensitivity of toehold switch-based detection systems.
Table 2: Performance Comparison of TEV Protease Systems
| Parameter | Wild-type TEV | uTEV1Δ | TEVp-C1 |
|---|---|---|---|
| Catalytic rate ((k_{cat})) | 0.18 s⁻¹ | Not specified | Not specified |
| Temporal resolution | >30 minutes | 60 seconds | Not specified |
| P1' residue specificity | G/S | G/S | Broadened specificity |
| Key advantage | High sequence specificity | Enhanced catalytic efficiency | Traceless cleavage |
| Primary limitation | Slow catalysis | Not specified | Not specified |
Experimental Protocol: Implementing TEV Protease Amplification in Toehold Switch Systems
Materials and Reagents
Table 3: Essential Research Reagent Solutions
| Reagent | Function | Specifications |
|---|---|---|
| uTEV1Δ protease | Signal amplification | Evolved TEV variant with S153N mutation |
| Toehold switch plasmid | RNA detection | Contains trigger-complementary region |
| pCRY-CIBN vector | Light-inducible control | CRY-CIBN photo-inducible protein pair |
| Transcription factor fusion | Reporter activation | Membrane-anchored TF with TEVcs |
| Citrine/mCherry reporters | Fluorescence readout | Normalization and activity measurement |
| Cell-free protein synthesis system | In vitro detection | PURExpress or similar |
Protocol: Integration of TEV Protease with Toehold Switches for Viral RNA Detection
Genetic Circuit Assembly
Construct Design: Clone the uTEV1Δ protease sequence downstream of a toehold switch-activated expression cassette, ensuring ribosome binding site optimization for your host system (E. coli recommended for initial validation).
Reporter Module: Incorporate a cleavable transcription factor (e.g., GAL4-EL222) fused to a plasma membrane anchor via a TEV cleavage site (TEVcs: ENLYFQ↓S/G). The cleaved TF should activate a detectable reporter (Citrine, GFP, or luciferase).
Control Elements: Include orthogonal regulatory elements such as the CRY-CIBN photo-inducible pair for system validation and tuning [52]. The mCherry fluorescent protein serves as an internal control for normalization.
System Validation and Optimization
Transformation and Expression: Transform the construct into BL21 E. coli or another appropriate expression host. Grow cultures at 37°C with appropriate antibiotics until OD₆₀₀ reaches 0.6.
Induction Protocol: Induce expression with 0.5 mM IPTG and incubate for 4-6 hours at 30°C to allow protein expression and maturation.
Trigger RNA Application: Add synthetic viral RNA triggers complementary to the toehold switch sequence. Use a concentration range of 1 nM to 1 μM for initial characterization.
Time-Course Monitoring: Measure fluorescence output (Citrine and mCherry) at 30-minute intervals for 6-8 hours using a plate reader or flow cytometry.
Signal Detection and Analysis
Signal Normalization: Calculate the Citrine/mCherry ratio for each sample to account for cell density and expression variability.
Kinetic Analysis: Determine the time to half-maximal activation (t₁/₂) and maximum fold-change over background.
Sensitivity Assessment: Perform dose-response curves with serial dilutions of trigger RNA to establish the limit of detection (LOD).
Diagram 1: TEV Protease Signal Amplification Pathway in Toehold Switch Systems
Troubleshooting and Optimization
Low Signal Amplification: Verify TEV protease activity using a fluorogenic peptide substrate (e.g., DABCYL-ENLYFQ↓S-GLUP-EDANS). Increase expression time or optimize ribosome binding site strength.
High Background: Incorporate additional transcriptional or translational insulation between circuit components. Consider using the TEVp S219N mutation to reduce self-cleavage and autoinactivation [53].
Slow Kinetics: Utilize the uTEV1Δ variant rather than wild-type TEV protease. For in vitro applications, consider adding enhancing additives such as glycerol or polyethylene glycol.
Application in Viral RNA Detection
The integration of engineered TEV protease systems with toehold switches creates a powerful platform for sensitive viral RNA detection. The two-stage amplification process—first at the RNA level through toehold switch activation, and second at the protein level through TEV protease-mediated reporter release—enables detection limits potentially reaching the attomolar range [54].
For viral diagnostics, this system can be adapted to cell-free expression platforms, allowing for point-of-care applications without specialized equipment [15]. The high sequence specificity of both toehold switches and TEV protease ensures minimal off-target activation, while the catalytic nature of TEV protease enables significant signal amplification from few initial trigger RNA molecules.
Diagram 2: Viral RNA Detection Workflow Using TEV-Amplified Toehold Switches
The strategic integration of engineered TEV protease systems with toehold switch technology creates a powerful signal amplification cascade that significantly enhances the sensitivity of viral RNA detection platforms. The uTEV1Δ variant, with its 27-fold improvement in signal-to-background ratio and capacity for minute-scale temporal resolution, addresses the fundamental limitation of slow catalysis in wild-type TEV protease [52]. This approach leverages the programmability of RNA-based detection with the catalytic amplification efficiency of engineered proteases, representing a promising direction for diagnostic applications requiring high sensitivity and specificity.
For researchers implementing this system, we recommend beginning with the uTEV1Δ variant in a well-characterized model system before adapting it to specific viral targets. The modular nature of both toehold switches and TEV protease systems enables straightforward customization for different diagnostic applications while maintaining the core signal amplification architecture described in this protocol.
Toehold switches are synthetic riboregulators that activate gene expression in response to a specific trigger RNA sequence through toehold-mediated strand displacement [1]. While these programmable RNA switches offer wide dynamic range and orthogonality, achieving high specificity against non-target RNAs and maintaining functionality amid viral mutations presents significant design challenges. Cross-reactivity occurs when switches respond to phylogenetically similar but non-target RNAs, compromising diagnostic accuracy and circuit reliability. Furthermore, the high mutation rates of RNA viruses can render detection elements obsolete as target sequences evolve, particularly in structural protein genes under selective pressure from host immunity [3]. This application note details evidence-based strategies and experimental protocols to overcome these specificity challenges, enabling robust toehold switch performance in viral detection systems.
Strategic Design Principles for Enhanced Specificity
Target Region Selection Criteria
Table 1: Guidelines for Target Region Selection in Viral Genomes
| Design Consideration | Recommended Approach | Rationale |
|---|---|---|
| Conservation Analysis | Target non-structural protein genes (e.g., Nsp2) over structural genes | Structural genes experience higher selective pressure from vaccination/infection, leading to more frequent mutations [3] |
| Cross-reactivity Avoidance | Perform BLAST analysis against human coronaviruses (HCoV-OC43, HCoV-229E, HCoV-NL63) and related viral genomes | Ensures trigger sequence is unique to target virus and minimizes false positives from co-circulating pathogens [3] |
| Secondary Structure Avoidance | Avoid pseudoknots between orfA and orfB genes, untranslated regions (UTRs), and regions with predicted local secondary structure | Unstructured regions facilitate more efficient strand displacement and toehold binding [3] |
| Mutation Resilience | Focus on regions with low mutation rates identified through genomic epidemiology databases | Designs remain functional across viral variants and emerging strains [3] |
Toehold Switch Architectural Design
The fundamental toehold switch architecture consists of several key domains that must be optimized for specificity:
- Toehold Domain: A 12-nucleotide single-stranded region that initiates binding with the trigger RNA [3]
- Stem Sequence: An 18-base pair stem that sequesters the ribosome binding site and start codon in the OFF state [1]
- Loop Region: Contains the ribosome binding site (RBS) and is typically 11 nucleotides in length [3]
- Start Codon Positioning: Located 6 nucleotides after the loop to ensure proper sequestration in the OFF state [3]
Unlike traditional riboregulators that rely on U-turn loop structures and RBS sequestration, toehold switches employ linear-linear interactions and sequester the region around the start codon, providing greater programmability and orthogonality [1]. This design enables forward engineering of switches with average dynamic ranges exceeding 400-fold activation [1].
Experimental Protocols for Specificity Validation
In Vitro Characterization of Toehold Switch Specificity
Materials Required:
- PURExpress In Vitro Protein Synthesis Kit (or similar CFPS system)
- DNA templates encoding toehold switches with T7 promoters
- Synthetic trigger RNA sequences (target and non-target)
- Fluorescent reporter plasmid (mNeonGreen or eGFP)
- Microplate reader for fluorescence detection
- Materials for RNA extraction and purification
Procedure:
Switch Design and Cloning:
- Design toehold switches using the STORM toehold switch generator software or manually with parameters from Table 1 [3]
- Clone switches into expression vectors containing T7 promoters and reporter genes (mNeonGreen is recommended over eGFP due to 3-5× higher intensity) [3]
- Verify sequences through Sanger sequencing before experimentation
Cell-Free Expression System Setup:
- Prepare reaction mixtures containing PURExpress cell-free system, DNA templates (10-20 nM), and supplemental energy sources
- Aliquot reactions into separate wells for testing with different trigger RNAs
- Add target and non-target trigger RNAs in a concentration series (e.g., 1 fM to 1 nM)
- Include no-trigger controls to assess baseline leakage
Specificity Assessment:
- Incubate reactions at 37°C for 4-6 hours with fluorescence measurements taken hourly
- Calculate ON/OFF ratios by comparing fluorescence with target trigger versus no-trigger control
- Assess cross-reactivity by testing against non-target triggers from related viruses or human transcripts
- Determine limit of detection using serial dilutions of target trigger RNA
Data Analysis:
- Normalize fluorescence readings to negative controls
- Calculate fold activation for each switch/trigger combination
- Apply threshold criteria (e.g., >10× higher response to target vs. non-target triggers) to determine specificity
Signal Amplification for Enhanced Sensitivity
Table 2: Comparison of Signal Amplification Strategies
| Amplification Method | Mechanism | Sensitivity Achieved | Advantages |
|---|---|---|---|
| TEV Protease System | Toehold activates TEV protease expression, which cleaves multiple quenched fluorescent substrates | Low femtomolar range (fM) | Eliminates need for upstream RNA amplification; modular design [3] |
| NASBA Pre-amplification | Isothermal amplification of target RNA prior to detection | 3 femtomolar (fM) | Established protocol; high sensitivity [3] |
| Dual Toehold Switches | Sequential activation of two toehold switches for CRISPR-based detection | Enhanced specificity through logic gating | Reduces false positives; enables complex circuit integration [7] |
TEV Protease Amplification Protocol:
Construct Design:
- Replace standard fluorescent reporter with coding sequence for tobacco etch virus (TEV) protease
- Design quenched fluorescent substrate with TEV cleavage site (e.g., ENLYFQ↓G)
Reaction Assembly:
- Combine CFPS system, toehold switch DNA, and target RNA trigger
- Include quenched fluorescent reporter substrate in the reaction mixture
- Run parallel reactions with non-target triggers as specificity controls
Signal Detection:
- Monitor fluorescence dequenching over time (typically 6-8 hours)
- Compare signal amplification factor to direct reporter systems
- Validate detection limit using diluted target RNA in biologically relevant matrices
Diagram 1: TEV protease signal amplification pathway for enhanced detection sensitivity.
Computational Approaches for Rational Design
Deep Learning for Function Prediction
Traditional thermodynamic models using NUPACK and ViennaRNA show limited predictive power for toehold switch function (R² = 0.04-0.15 for ON/OFF ratios) [15]. Deep neural networks (DNNs) trained on large-scale datasets (e.g., 91,534 switches spanning 23 viral genomes) significantly outperform these methods (R² = 0.43-0.70) [15].
Implementation Protocol:
Data Preparation:
- Curate sequence dataset of toehold switches with experimental performance metrics
- Include diverse viral targets and human transcription factors for broad applicability
- Balance dataset to address skew toward low-signal variants in OFF states
Model Training:
- Implement DNN architecture with attention mechanisms for sequence analysis
- Train on nucleotide sequences rather than derived thermodynamic parameters
- Utilize VIS4Map (Visualizing Secondary Structure Saliency Maps) to interpret learned patterns
Design Optimization:
- Generate candidate switches using trained model
- Prioritize designs with predicted high ON/OFF ratios and low cross-reactivity
- Validate top candidates experimentally before full-scale synthesis
Diagram 2: Computational workflow for rational toehold switch design.
Advanced Applications: CRISPR Integration
The integration of toehold switches with CRISPR systems creates intelligent guide RNAs (IngRNAs) that respond to specific cellular triggers [7]. These systems employ dual toehold switches for conditional activation of CRISPR function, enhancing specificity through logic-gated operations.
IngRNA Implementation Protocol:
Construct Design:
- Design IngRNA with two toehold domains and sequestered CrRNA sequence
- Position first toehold in upper loop for trigger RNA binding
- Ensure second toehold becomes accessible only after initial trigger binding
Validation in Cellular Systems:
- Test IngRNA function in bacterial (BL21 E. coli) and mammalian (HEK293T) cells
- Measure target gene regulation (e.g., luciferase repression) with and without trigger RNA
- Assess off-target effects using RNA sequencing or targeted PCR
Specificity Controls:
- Include non-cognate trigger sequences to validate switching specificity
- Test in complex biological samples to assess performance in realistic conditions
Research Reagent Solutions
Table 3: Essential Materials for Toehold Switch Development
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cell-Free Expression Systems | PURExpress, homemade E. coli S30 extracts | In vitro transcription/translation for rapid switch characterization [3] |
| Fluorescent Reporters | mNeonGreen, eGFP, luciferase | Quantitative measurement of switch activation; mNeonGreen offers 3-5× higher intensity than eGFP [3] |
| Signal Amplification Components | TEV protease, quenched fluorescent substrates | Enhanced sensitivity without target pre-amplification; enables femtomolar detection [3] |
| High-Fidelity Polymerases | Phusion, Herculase II | PCR amplification of toehold switch constructs with minimal error rates [3] [7] |
| Computational Design Tools | NUPACK, ViennaRNA, STORM toehold generator | Prediction of RNA secondary structure and switch performance [3] [15] |
| Cloning Systems | pET28a, pTargetF, pUC19-based vectors | Expression of toehold switches and reporter genes in prokaryotic systems [3] [7] |
Overcoming specificity challenges in toehold switch design requires integrated computational and experimental approaches. Strategic target selection focusing on conserved, low-mutation regions with minimal secondary structure provides the foundation for specific viral detection. Advanced signal amplification strategies like the TEV protease system enable clinically relevant sensitivity without pre-amplification, while deep learning models dramatically improve predictive design capabilities. The integration of toehold switches with CRISPR systems further expands their application space through logic-gated operation. By implementing the protocols and design principles outlined in this application note, researchers can develop robust toehold switch-based detection systems that maintain specificity across viral variants and minimize cross-reactivity with related pathogens.
In the realm of synthetic biology and molecular diagnostics, toehold switches have emerged as powerful programmable riboregulators for detecting specific RNA sequences, including viral pathogens. These synthetic biological elements operate through a mechanism known as toehold-mediated strand displacement (TMSD), where a trigger RNA binds to a complementary "toehold" region on the switch, initiating a structural rearrangement that activates gene expression [55] [56]. The kinetics of this displacement reaction—the speed and efficiency with which the switch transitions from an OFF to an ON state—are critically dependent on the structural attributes of the toehold domain itself. For researchers developing diagnostic platforms for viral RNA detection, mastering the relationship between toehold design and kinetic performance is essential for creating sensitive, rapid, and reliable biosensors [12] [11]. This Application Note examines the fundamental principles and practical considerations for optimizing toehold domain parameters to achieve desired kinetic outcomes in viral detection systems.
Theoretical Foundation of Toehold-Mediated Strand Displacement
Kinetic Mechanism
Toehold-mediated strand displacement is a fundamental reaction in dynamic DNA/RNA nanotechnology. The process follows a three-step mechanism:
- Toehold Binding: The invading trigger RNA strand hybridizes to the single-stranded toehold domain of the switch.
- Branch Migration: Through a random walk process, the trigger RNA displaces the incumbent strand from the switch complex.
- Strand Dissociation: The displaced strand detaches, resulting in a stable trigger-switch complex and activation of the reporter gene [56].
The initial toehold binding is typically the rate-limiting step, making the structural and thermodynamic properties of the toehold domain paramount to the overall reaction kinetics. The reaction follows a second-order kinetic model: I + TC → k_TMSD IT + C, where the rate constant k_TMSD can reach up to ~10⁶ M⁻¹s⁻¹ [56].
Key Performance Metrics
For viral detection applications, toehold switch performance is evaluated using several key metrics:
- Activation Kinetics (k_TMSD): The rate constant for the strand displacement reaction, determining how quickly a signal is generated upon trigger recognition.
- ON/OFF Ratio: The fold-change in reporter gene expression (e.g., fluorescence, colorimetric signal) between the triggered (ON) and untriggered (OFF) states. A high ratio is crucial for minimizing background signal and maximizing detection sensitivity [55].
- Specificity: The ability to discriminate between the target trigger RNA and non-target sequences, including single-nucleotide variants, which is vital for accurate viral strain identification [12].
Optimizing Toehold Domain Parameters
The following protocols and data analyses provide a systematic approach for tuning toehold domains to achieve optimal kinetic performance.
Protocol: Determining Optimal Toehold Length
Objective: To empirically determine the optimal toehold length that maximizes the activation rate and ON/OFF ratio for a specific viral RNA target.
Materials:
- NUPACK or ViennaRNA software for in silico design [55] [11]
- DNA oligonucleotides for toehold switch and trigger RNA synthesis
- In vitro transcription kit (e.g., T7 High Yield RNA Synthesis Kit) [7]
- Cell-free protein synthesis system (e.g., NEBExpress)
- Reporter system (e.g., GFP for fluorescence, LacZ/CPRG for colorimetry) [11]
- Plate reader or spectrophotometer
Method:
- Design: Design a series of toehold switches targeting a conserved region of the viral genome (e.g., the HC-Pro or CP genes for TuMV) [11]. Vary the toehold length from 0 to 12 nucleotides (nt), keeping all other sequence elements constant.
- Computational Validation: Use NUPACK to predict the minimum free energy (MFE) structures of each switch. Calculate the free energy of the OFF state (ΔGoff) and the free energy change upon trigger binding (ΔΔG). Select designs with ΔGoff between -5 and -15 kcal/mol for sufficient stability and ΔΔG between -10 and -20 kcal/mol for effective triggering [55].
- Synthesis: Synthesize the DNA templates and transcribe the toehold switch RNAs and trigger RNAs in vitro.
- Testing: For each toehold switch variant, combine the following in a cell-free reaction:
- Toehold switch RNA (e.g., 50 nM)
- Trigger RNA (e.g., a concentration gradient from 0.1 nM to 100 nM)
- Cell-free expression machinery
- Reporter gene construct
- Incubation: Incubate the reactions at 37°C and monitor reporter output over time (e.g., fluorescence every 5 minutes for 2 hours).
- Data Analysis: Calculate the apparent activation rate constant (k_obs) from the initial linear phase of signal generation. Determine the ON/OFF ratio as the maximum signal in the presence of a saturating trigger concentration divided by the signal in the absence of trigger.
Table 1: Expected Kinetic Parameters vs. Toehold Length
| Toehold Length (nt) | Theoretical k_TMSD (M⁻¹s⁻¹) | Expected ON/OFF Ratio | Key Considerations |
|---|---|---|---|
| 0-2 | < 10³ | Low (< 5) | Very slow kinetics; insufficient to drive efficient activation. |
| 3-5 | 10³ - 10⁴ | Moderate (5-50) | Good for balancing specificity and speed; often optimal. |
| 6-8 | ~10⁵ - 10⁶ | High (50-500) | Often the "sweet spot" for maximal rate and high output [56]. |
| 9-12 | ~10⁶ (plateau) | High (potential for increased leakiness) | Maximal kinetics, but may suffer from increased non-specific activation or sensor instability [55]. |
Protocol: Tuning Kinetics via Sequence Composition and GC Content
Objective: To investigate the effect of toehold sequence composition and GC content on strand displacement kinetics and specificity.
Materials: (Same as Protocol 3.1, with the addition of non-complementary or mismatched trigger RNAs for specificity testing.)
Method:
- Design: For a fixed toehold length (e.g., 6 nt), design multiple sequences where the GC content varies from 0% to 100%. Ensure the sequences do not form stable secondary structures internally or with non-target regions of the switch.
- Stability Prediction: Use ViennaRNA or NUPACK to calculate the free energy of toehold-trigger binding (ΔGbinding). Note that lower (more negative) ΔGbinding generally correlates with faster binding kinetics.
- Synthesis and Testing: Synthesize and test the switch variants as described in Protocol 3.1.
- Specificity Assessment: Test each switch against:
- The perfectly complementary trigger RNA.
- Triggers with single-nucleotide polymorphisms (SNPs) relevant to viral strain differentiation.
- A non-target RNA sequence. Use a single, clinically relevant concentration of trigger for these comparisons.
Table 2: Impact of Sequence Composition on Toehold Switch Performance
| GC Content | ΔG_binding (kcal/mol, approx.) | Impact on Kinetics | Impact on Specificity | Recommendation |
|---|---|---|---|---|
| Low (0-33%) | > -5 | Slow | High | Useful when high specificity is critical and speed is secondary. |
| Medium (40-60%) | -5 to -10 | Moderate to Fast | High | Optimal for most applications; provides a balance of speed and fidelity [55]. |
| High (67-100%) | < -10 | Very Fast | Reduced | Can lead to increased off-rates and non-specific binding; avoid >80% GC [55]. |
Advanced Strategy: Genetic Algorithms for Multi-Parameter Optimization
Objective: To simultaneously optimize multiple conflicting parameters (e.g., kinetics, stability, specificity) using a computational genetic algorithm.
Method:
- Define Fitness Function: Create a fitness function that weights key performance metrics, such as:
Fitness = w1 * (ON/OFF Ratio) + w2 * (k_TMSD) - w3 * (Leakiness) - w4 * (Dimer_Tendency)whereware weighting factors determined by project goals. - Generate Initial Population: Use NUPACK to generate an initial population of toehold switch sequences under defined constraints [55].
- Iterate: Allow the population to evolve over generations through crossover and mutation events. In each generation, evaluate the fitness of each sequence using the defined function and computational predictions (e.g., from NUPACK and ViennaRNA) [55].
- Selection: Select the highest-performing sequences for experimental validation.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Toehold Switch Development and Validation
| Reagent / Tool | Function / Purpose | Example Product / Source |
|---|---|---|
| NUPACK | Computational design and MFE prediction of nucleic acid sequences [55] [56]. | nupack.org |
| ViennaRNA Package | Prediction of RNA secondary structure and folding thermodynamics [11]. | rna.tbi.univie.ac.at |
| T7 High Yield RNA Synthesis Kit | In vitro transcription of toehold switch and trigger RNAs [7]. | New England Biolabs |
| Cell-Free Protein Synthesis System | Expression of reporter protein upon toehold switch activation without using live cells [11]. | NEBExpress, PURExpress |
| Colorimetric Reporter (LacZ/CPRG) | Visual readout of sensor activation; suitable for field applications [11]. | Chlorophenol Red-β-D-galactopyranoside |
| Fluorescent Reporter (GFP/mCherry) | Quantitative measurement of kinetics and ON/OFF ratios via plate reader [55] [7]. | Green Fluorescent Protein variants |
Workflow Visualization
The following diagram illustrates the complete experimental and computational workflow for developing and optimizing a toehold switch for viral detection, integrating the protocols described above.
Workflow for Toehold Switch Optimization
The rational design of the toehold domain is a critical determinant in the performance of toehold switch-based biosensors. By systematically varying the toehold length and fine-tuning the sequence composition, researchers can directly influence the activation kinetics, sensitivity, and specificity of their diagnostic systems. The protocols outlined herein provide a robust framework for this optimization process. Integrating computational design with empirical validation, as part of an iterative design-build-test cycle, significantly accelerates the development of highly effective toehold switches. Mastering these principles is key to advancing the field of RNA-based diagnostics and deploying rapid, accurate, and field-deployable tests for viral pathogens.
Within the field of viral diagnostics using synthetic biology, toehold switch sensors have emerged as a powerful tool for the specific detection of pathogen RNA [8]. A significant challenge in deploying these biosensors, particularly for point-of-care applications, lies in handling the complex matrices of real-world samples. The choice between using crude or purified RNA extracts directly impacts the assay's sensitivity, speed, cost, and feasibility in resource-limited settings. Crude extracts often contain inhibitors that can compromise sensor performance, while traditional RNA purification adds time, cost, and requires specialized equipment [57]. This application note details experimental protocols and data for using toehold switches for viral RNA detection in both crude and purified RNA extracts, providing a framework for researchers to develop robust field-deployable diagnostics.
Performance Comparison & Quantitative Data
Toehold switch sensors have been successfully validated for detecting plant viral pathogens such as Turnip mosaic virus (TuMV) and Potato Virus Y (PVY) in both purified and crude RNA extracts [58] [57]. The performance characteristics, however, vary between the two methods.
Table 1: Performance Metrics of Toehold Switches with Purified vs. Crude RNA Extracts
| Performance Metric | Purified RNA Extracts | Crude RNA Extracts |
|---|---|---|
| Detection Limit | < 10 fM (with 90 min assay) [58] | Comparable to commercial systems when optimized [57] |
| Time to Result | ~40 min (for 1 pM detection) [58] | ~3 hours (total, including processing) [58] |
| Key Advantage | Maximum sensitivity; reduced biochemical background [58] | Rapid sample preparation; suitable for field deployment [58] [57] |
| Main Limitation | Time-consuming and expensive purification process; requires lab equipment [57] | Potential for assay inhibition; may require optimized lysates [57] [59] |
| Specificity | High (no cross-reactivity with Cucumber Mosaic Virus) [58] | Successfully demonstrated for PVY [57] |
Detailed Experimental Protocols
Protocol A: Viral RNA Detection Using Purified RNA Extracts
This protocol is adapted from studies detecting TuMV in Pseudostellaria heterophylla and uses Nucleic Acid Sequence-Based Amplification (NASBA) coupled with a toehold switch sensor for high sensitivity [58].
Step 1: RNA Purification
- Extract total RNA from infected plant tissue (e.g., leaves) using a commercial kit or a standard phenol-chloroform method.
- Quantify RNA using a spectrophotometer (e.g., Nanodrop). An A260/A280 ratio of ~1.8-2.0 and an A260/A230 ratio of >2.0 are indicative of pure RNA [60].
- Optional Quality Control: Use an external standard RNA added to the sample prior to lysis to evaluate the yield, degradation, and presence of enzyme inhibitors in the extracted RNA [60].
Step 2: Isothermal Amplification (NASBA)
- Prepare the NASBA reaction mixture on ice:
- Components: 1x NASBA buffer, 15% DMSO, 1 M Betaine, 40 U of T7 RNA Polymerase, 8 U of AMV Reverse Transcriptase, 0.2 U of RNase H, 2 mM each dNTP, 2 mM each NTP, and 15 mM MgCl₂ [58] [59].
- Primers: Include forward primers containing a T7 RNA polymerase promoter sequence [58].
- Template: Add 1-10 µL of the purified RNA extract.
- Incubate the reaction at 41°C for 90 minutes [58]. This step simultaneously amplifies the target RNA and produces complementary triggers for the toehold switch.
- Prepare the NASBA reaction mixture on ice:
Step 3: Toehold Switch Detection
Protocol B: Viral RNA Detection Using Crude RNA Extracts
This protocol leverages low-cost, locally producible cell extracts and simplifies sample processing for field application, as demonstrated for PVY detection [57].
Step 1: Rapid Crude Extract Preparation
- Grind a small amount of plant tissue (e.g., ~100 mg) in a microtube with 200 µL of a simple lysis buffer (e.g., containing 0.5% Triton X-100) [59].
- Centrifuge the lysate briefly (e.g., 1-2 minutes at >10,000 × g) to pellet debris.
- Transfer the supernatant, which contains the crude RNA, to a new tube. The supernatant can be used directly or with minimal dilution in the subsequent amplification step [57].
Step 2: Coupled Amplification & Detection
- Prepare a one-pot reaction mixture containing:
- Amplification Components: RT-RPA or NASBA reagents, similar to Protocol A, but optimized for compatibility with crude lysates [57] [59].
- Cell-Free System: Use an optimized, low-cost E. coli cell extract. To enhance performance, extracts can be pre-engineered using CRISPRi to knock down nucleases, improving the stability of linear DNA templates [57].
- Toehold Switch & Reporter: DNA template for the switch and the necessary components for the reporter output (e.g., lacZα and substrate).
- Add the crude RNA extract supernatant directly to the reaction mixture.
- Incubate the single-pot reaction at a constant temperature (37-41°C for maximum speed or room temperature for equipment-free operation) for approximately 3 hours and observe the readout [58] [59].
- Prepare a one-pot reaction mixture containing:
Workflow Visualization
The following diagram illustrates the key decision points and steps in the two protocols.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Toehold Switch-Based RNA Detection
| Reagent / Material | Function / Explanation | Example Protocols / Notes |
|---|---|---|
| Toehold Switch Plasmid | De novo designed riboswitch; binds trigger RNA to activate translation of a reporter gene [58] [8]. | Designed to be complementary to a specific region of the target viral RNA (e.g., TuMV CP gene) [58]. |
| Cell-Free Protein Synthesis (CFPS) System | In vitro platform for transcription and translation; enables sensor operation outside living cells [57] [59]. | Use commercial PURE system for high performance or low-cost, in-house E. coli extracts for decentralization [57]. |
| Reporter System | Generates a measurable signal (colorimetric, fluorescent) upon toehold switch activation [21] [59]. | lacZα for blue color with X-gal; sfGFP for fluorescence [59]. sfGFP offers faster, brighter signals [21]. |
| Isothermal Amplification Reagents | Amplifies target RNA to detectable levels without complex thermocycling, crucial for sensitivity [58]. | NASBA (for RNA amplification) or RT-RPA. Includes enzymes like T7 RNA Polymerase and Reverse Transcriptase [58] [59]. |
| Low-Cost Cell Extracts | Crude E. coli lysates prepared in-house to drastically reduce cost and eliminate cold-chain dependence [57]. | Can be optimized via CRISPRi to knock down nucleases, enhancing DNA template stability for better performance [57]. |
The choice between crude and purified RNA extracts for toehold switch-based detection involves a direct trade-off between ultimate sensitivity and operational practicality. Purified RNA protocols are the gold standard for laboratory-based research where detection limits are paramount. However, for field-deployable diagnostics, animal health monitoring, and resource-limited settings, the use of crude extracts with optimized, low-cost cell-free systems presents a revolutionary and viable path forward. By adopting the protocols and optimization strategies outlined here, researchers can advance the development of robust, affordable, and rapid molecular diagnostics that function reliably in complex real-world matrices.
The imperative for room-temperature stable reagents is a cornerstone of deployable diagnostic and therapeutic technologies. For research utilizing toehold switches for viral RNA detection, lyophilization (freeze-drying) presents a transformative strategy to enhance the stability and shelf-life of sensitive biological components, thereby facilitating their global distribution and application. These Application Notes and Protocols provide a detailed, practical framework for the development and optimization of lyophilized formulations, with a specific focus on stabilizing the complex biochemical reagents inherent to toehold switch systems. We summarize critical quantitative parameters, provide step-by-step experimental methodologies, and outline essential quality control measures to guide researchers and drug development professionals in creating stable, room-temperature formulations.
Toehold switch systems for viral RNA detection represent a powerful and programmable diagnostic technology. However, their components, including specialized enzymes, reporter molecules, and the RNA switches themselves, are inherently labile, often requiring cold-chain logistics for storage and shipping. This dependency creates significant barriers to their widespread use, particularly in resource-limited settings. Lyophilization addresses this challenge by removing water from the product under vacuum and low temperature, converting it into a stable solid cake. This process dramatically reduces molecular mobility and halts degradation pathways such as hydrolysis, enabling long-term storage at refrigerated or even room temperatures [61] [62]. For research and commercial applications involving toehold switches, a well-designed lyophilized formulation ensures that critical reagents retain their functionality—such as the switch's ability to undergo conformational change and initiate translation upon target binding—from manufacturing through to end-use, without the burden of a cold chain [63].
Core Principles of Lyophilization Formulation
A successful formulation is predicated on a systematic, data-driven approach grounded in Quality by Design (QbD) principles [61]. The process involves several key stages:
- Pre-formulation and Developability Assessment: The first step is a thorough characterization of the molecule's vulnerabilities. For a toehold switch system, this involves identifying the specific stresses—thermal, mechanical, or chemical—that trigger degradation pathways like aggregation or loss of function. This foundational data informs the entire formulation strategy [61] [63].
- Critical Temperature Determination: A fundamental parameter is the formulation's collapse temperature (
T_c), the maximum temperature the product can withstand during primary drying without losing its structural integrity.T_cis closely related to the glass transition temperature of the maximally freeze-concentrated solute (T_g'). To produce a pharmaceutically elegant and stable cake, the product temperature must be maintained belowT_cduring primary drying [64] [65]. This temperature is empirically determined using techniques like Freeze-Dry Microscopy. - Excipient Engineering: Excipients are not inert fillers; they are functional ingredients designed to protect the active molecule. A rational combination is typically required [61] [66]:
- Cryoprotectants (e.g., sucrose, trehalose) protect the molecule during the freezing phase.
- Lyoprotectants stabilize the molecule during drying by replacing water molecules that normally form hydrogen bonds with the biologic, thus preserving its native structure in the solid state.
- Bulking Agents (e.g., mannitol, glycine) provide structural support to the lyophilized cake, ensuring elegant appearance and friability. Crystalline bulking agents like mannitol can also raise the eutectic temperature of the formulation, allowing for a more efficient drying cycle [61] [65].
Quantitative Formulation Data
The selection and ratio of excipients are critical for achieving both stability and process efficiency. The following table summarizes key data on common excipient classes and optimized combinations from relevant literature.
Table 1: Lyophilization Excipients and Their Functional Roles
| Excipient Class | Example Compounds | Primary Function | Key Quantitative Findings |
|---|---|---|---|
| Disaccharide Cryo/Lyoprotectants | Sucrose, Trehalose | Stabilize biological structures during freezing and drying; form an amorphous glassy matrix. | A mixed lyoprotectant of Sucrose:Trehalose:Mannitol (5:5:1 w/w/w) increased system collapse temperature, enabling a shorter (8-18 hr) lyophilization cycle while maintaining particle integrity [66]. |
| Bulking Agents | Mannitol, Glycine | Provide crystalline structure and cake elegance; prevent blow-out. | Mannitol crystallizes upon freezing, providing a rigid cake structure. However, its crystallization must be controlled to avoid compromising the amorphous stabilizer matrix [61] [65]. |
| Buffers | Tris, Histidine, Succinate | Control pH during freezing and in the final reconstituted product. | Buffer salts can crystallize during freezing, leading to dramatic pH shifts (≥3 pH units). Non-crystallizing buffers are often preferred for sensitive biologics [65]. |
| Surfactants | Polysorbate 20, Polysorbate 80 | Mitigate interfacial stresses (ice-liquid, air-liquid) that can cause protein aggregation or particle clumping. | Used at low concentrations (e.g., 0.01%-0.1%) to prevent surface-induced degradation of sensitive nanoparticles and proteins [62]. |
Table 2: Critical Quality Attributes (CQAs) for a Lyophilized Toehold Switch Reagent
| Quality Attribute | Target Profile | Analytical Method |
|---|---|---|
| Residual Moisture | 1% - 3% | Karl Fischer Titration |
| Reconstitution Time | < 60 seconds | Visual and manual timing |
| Cake Appearance | White, friable, uniform cake with no collapse | Visual inspection |
| Particle Size / PDI | Maintained pre-lyophilization distribution (e.g., PDI < 0.2) | Dynamic Light Scattering (DLS) |
| mRNA Integrity | RNA Integrity Number (RIN) > 8.0 | Capillary Electrophoresis (e.g., Bioanalyzer) |
| Functional Assay (Potency) | > 90% recovery of diagnostic signal (e.g., fluorescence upon activation) | In vitro toehold switch activation assay |
Detailed Experimental Protocols
Protocol 1: Pre-lyophilization Formulation Screening and Preparation
Objective: To identify a stable preliminary formulation and prepare it for the lyophilization cycle.
Materials:
- Toehold switch reagent (e.g., complete master mix with enzymes and switches)
- Excipients: Sucrose, Trehalose, Mannitol (USP grade)
- Buffer: e.g., 10 mM Tris-HCl, pH 7.5
- Surfactant: e.g., Polysorbate 20
- Sterile, nuclease-free water
- 3 mL Type I glass lyophilization vials
- Lyophilization stoppers and caps
Methodology:
- Formulation Screening: Prepare multiple small-scale (e.g., 1 mL) formulations based on a Design of Experiments (DoE) approach. A suggested starting point is a mixture of 5% sucrose, 5% trehalose, and 1% mannitol (w/v) in 10 mM Tris-HCl buffer, pH 7.5, with 0.01% Polysorbate 20 [66].
- Stress Testing: Subject each formulation to defined stress conditions (e.g., 3-5 freeze-thaw cycles, agitation at 300 rpm for 1 hour, incubation at 25°C for 24 hours).
- CQA Analysis: After stress testing, analyze all formulations for key CQAs from Table 2, with a primary focus on functional assay performance and particle size/PDI.
- Bulk Formulation: Based on screening results, prepare the selected formulation at the required bulk volume. Filter sterilize the solution using a 0.22 µm PES filter.
- Vial Filling: Aseptically fill the formulated solution into clean, sterile 3 mL lyophilization vials. Use a target fill volume that provides a reasonable cake depth (e.g., 1 mL in a 3 mL vial). Partially stopper the vials with lyophilization stoppers.
Protocol 2: Optimized Lyophilization Cycle Development
Objective: To execute a lyophilization cycle designed to preserve the structural and functional integrity of the toehold switch reagent.
Materials:
- Pilot-scale lyophilizer (e.g., with shelf area of 0.5 - 1 m²)
- Partially stoppered vials from Protocol 1
- Thermocouples (if available for process monitoring)
Methodology: The lyophilization process consists of three distinct stages, as visualized in the following workflow:
Lyophilization Cycle Parameters:
- Freezing:
- Primary Drying (Sublimation):
- Reduce the chamber pressure to 100 mTorr.
- Increase the shelf temperature to -25°C (ensuring this is below the formulation's
T_c). Ramp slowly at 0.5°C/min. - Hold at these conditions for approximately 20 hours. This is the longest phase, where sublimation of ice occurs. The endpoint can be determined by a pressure rise test or a tunable diode laser absorption spectroscopy (TDLAS) system, indicating that all ice has sublimed [64] [67].
- Secondary Drying (Desorption):
- Back-filling and Stoppering:
- After secondary drying, break the vacuum by back-filling the chamber with dry, sterile nitrogen gas to an atmospheric pressure of 700-800 mTorr.
- Fully stopper the vials within the chamber using the hydraulic stoppering mechanism.
Quality Control and Stability Assessment
Objective: To verify that the lyophilized product meets all pre-defined CQAs and possesses the required shelf-life.
Methodology:
- Post-Lyophilization Analysis: Immediately after lyophilization, test vials for the CQAs listed in Table 2. This includes checks for cake appearance, residual moisture, reconstitution time, and most critically, the functional activity of the toehold switch reagent.
- Real-time Stability Studies: Place the final lyophilized product on stability studies at the intended storage temperature (e.g., 2-8°C, 25°C/60%RH). Sample vials at predetermined timepoints (e.g., 0, 1, 3, 6, 12, 24 months) and repeat the full panel of CQA tests. The data generated here is essential for defining the product's shelf-life and is a regulatory requirement for commercial products [61] [65].
- Accelerated Stability Studies: Expose the product to accelerated conditions (e.g., 40°C/75%RH) to rapidly screen for major instability issues and predict long-term stability trends.
The entire development and quality control pathway, from formulation to a stable product, is summarized below:
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Lyophilization Development
| Item Category | Specific Examples | Function / Application Note |
|---|---|---|
| Stabilizing Excipients | Sucrose (USP), Trehalose (Dihydrate, USP), D-Mannitol (USP) | Form the stabilizing amorphous cake matrix (sugars) and provide structural rigidity (mannitol). Source from GMP-grade suppliers for clinical applications. |
| Analytical Instruments | Freeze-Dry Microscope, Differential Scanning Calorimeter (DSC), Karl Fischer Titrator, Dynamic Light Scatter (DLS) | Determine critical formulation temperatures (T_c, T_g'), measure residual moisture, and assess particle size distribution pre- and post-lyophilization. |
| Lyophilization Equipment | Laboratory-scale Freeze Dryer (e.g., from SP Scientific, Millrock Technology) | For cycle development and small-batch production. Ensure the unit has controllable shelf temperature, vacuum pressure, and process analytics (e.g., Pirani gauge). |
| Primary Packaging | 3 mL Type I Glass Lyophilization Vials, 13 mm Lyophilization Stoppers (e.g., West 4432/50) | Vials must withstand thermal and pressure stress. Stoppers must allow for water vapor transmission during drying and provide an airtight seal after stoppering. |
| Functional Assay Kits | Fluorescent RNA/DNA Quantitation Kits, Capillary Electrophoresis Systems (e.g., Agilent Bioanalyzer), In Vitro Transcription/Translation Kits | Used for potency testing to ensure the lyophilized toehold switch reagent retains its biological activity and detection sensitivity. |
Benchmarking Success: Validation Frameworks and Comparative Analysis with Gold Standards
Toehold switches are a class of de-novo-designed prokaryotic riboregulators that activate gene expression in response to cognate RNA triggers with arbitrary sequences [1]. These synthetic RNA devices have emerged as powerful tools for molecular detection due to their high orthogonality, programmability, and wide dynamic range, routinely enabling modulation of protein expression over two orders of magnitude [1]. In viral detection applications, toehold switches can be programmed to recognize specific pathogen RNA sequences, triggering a measurable signal output upon hybridization. The predictability of Watson-Crick base pairing and the availability of sophisticated computational design tools have positioned toehold switches as transformative components in diagnostic platforms, particularly for detecting viral RNAs from pathogens such as SARS-CoV-2 [12].
The performance of RNA-based biosensors is heavily influenced by the often-overlooked interplay between the sensor and its target RNA, where binding kinetics, secondary structure, and accessibility dictate function [12]. Establishing robust analytical performance parameters, specifically the Limit of Detection (LOD) and dynamic range, is therefore critical for deploying toehold switches in reliable diagnostic applications. The fundamental rules governing RNA-RNA interactions—specifically the structure-function relationships that determine sensor performance—remain poorly understood, necessitating systematic characterization approaches [12]. This protocol details the methodologies for quantitatively establishing these essential performance characteristics within the context of viral RNA detection research.
Theoretical Foundations of Performance Parameters
Defining Limit of Detection (LOD) and Limit of Quantification (LOQ)
In diagnostic procedures, the Limit of Detection (LOD) and Limit of Quantification (LOQ) are among the most critical performance parameters describing the minimum amount of target that can be reliably detected and quantified [68]. The Clinical Laboratory Standards Institute (CLSI) defines LOD as "the lowest amount of analyte in a sample that can be detected with (stated) probability, although perhaps not quantified as an exact value" [68]. In practice, LOD represents the lowest concentration at which a positive signal can be distinguished from background noise with a specified confidence level, typically 95%.
The LOQ is defined as "the lowest amount of measurand in a sample that can be quantitatively determined with stated acceptable precision and stated, acceptable accuracy, under stated experimental conditions" [68]. While LOD establishes presence/absence, LOQ defines the threshold for reliable quantitative measurement. For toehold switch applications, these parameters determine the clinical utility for early pathogen detection where viral load may be minimal.
Understanding Dynamic Range
Dynamic range refers to the concentration interval over which the analytical method provides quantitative results with acceptable accuracy and precision [1]. For toehold switches, this is typically reported as the ratio between the maximum and minimum quantifiable target concentrations, often expressed as the ON/OFF ratio—the fold-change in output signal between the triggered and basal states [1] [15]. Engineered toehold switches have demonstrated average dynamic ranges above 400, a performance level typically reserved for protein-based transcriptional regulators [1]. This wide dynamic range enables detection across clinically relevant viral load concentrations.
Table 1: Key Analytical Performance Parameters for Toehold Switches
| Parameter | Definition | Importance in Viral Detection |
|---|---|---|
| Limit of Detection (LOD) | Lowest viral RNA concentration detectable with stated probability | Determines earliest possible infection detection |
| Limit of Quantification (LOQ) | Lowest viral RNA concentration quantifiable with acceptable precision | Enables viral load monitoring for disease progression |
| Dynamic Range | Range between minimum and maximum quantifiable concentrations | Allows detection across various infection stages |
| ON/OFF Ratio | Fold-change between triggered and basal signal states | Indicates switch performance and signal-to-noise ratio |
Experimental Protocol for LOD and Dynamic Range Determination
Toehold Switch Design and Construction
The design of high-performance toehold switches requires careful consideration of both the sensor and target RNA structures. Follow this standardized procedure to generate functional toehold switches for viral RNA detection:
Switch Architecture Selection: Utilize the second-generation toehold-switch design (tsgen2, series A) to minimize sequence variability in regions contributing to secondary structure [12]. This architecture features a conserved top part of the switch, with the target RNA unwinding only six base pairs into the stem.
Target Site Identification: Scan the target viral genome for appropriate trigger binding sites using computational tools. Begin designs starting from index 21 of the RNA sequence to avoid putative cryptic ribosome binding sites [12]. For a target like SARS-CoV-2 RNA, evaluate all possible toehold switches across the target sequence.
Computational Validation: Employ RNA secondary structure prediction tools such as NUPACK [13] or ViennaRNA [13] to model switch-transcript interactions. Calculate minimum free energy (MFE), ideal ensemble defect (IED), and native ensemble defect (NED) parameters to evaluate design stability [15].
Sequence Optimization: Apply machine learning frameworks like Toehold-VISTA when possible, which integrates biophysical modeling of both sensor and target RNAs with partial least squares discriminant analysis (PLS-DA) to capture key determinants of RNA sensor performance [12].
Oligonucleotide Synthesis: Design template DNA oligos to contain a 5' T7 promoter sequence and a common 3' sequence featuring a 21-nt linker and the first 9 nt of the reporter gene (e.g., GFP) [12].
Plasmid Construction: Clone toehold switches into appropriate expression vectors. A two-plasmid system can be utilized to transcribe the target RNA in trans to the OFF-state switch RNA on pColE1 and pColA plasmids, respectively [12].
Toehold Switch Design Workflow
Establishing a Standard Curve for Quantification
Accurate quantification of toehold switch performance requires establishment of a standard curve using samples of known concentration:
Standard Preparation: Prepare a dilution series of synthetic target viral RNA covering the expected detection range. For comprehensive characterization, use a 2-fold dilution series covering a range from 1 to 2048 molecules per reaction volume [68].
Replication Scheme: Analyze each standard concentration in multiple replicates to ensure statistical robustness. Include at least 64 replicates per concentration, with additional replicates (e.g., 128) for the most diluted samples near the expected detection limit [68].
Assay Execution: Perform the toehold switch activation assay with all standard concentrations using consistent reaction conditions. For cell-free systems, use platforms like PURExpress with standardized incubation conditions [15].
Signal Detection: Measure output signals appropriate to your reporter system:
- For fluorescent reporters (e.g., GFP): Measure fluorescence intensity with a plate reader
- For colorimetric outputs: Measure absorbance at appropriate wavelengths
- For luminescent reporters: Measure luminescence intensity
Curve Fitting: Plot the measured signal against the logarithm of the target concentration and perform regression analysis to generate a standard curve. The curve should display a linear relationship in the quantitative range.
Determining Limit of Detection (LOD)
For toehold switch systems, LOD determination requires a probabilistic approach rather than conventional linear methods, as the response is logarithmic rather than linear [68]. Follow this statistical method:
Sample Preparation: Prepare a dilution series of target RNA focusing on the low concentration range expected near the detection limit. Include a minimum of 5-7 different concentrations with multiple replicates at each level (minimum 20-30 replicates per concentration) [68].
Experimental Runs: Conduct toehold switch activation assays with all dilution levels using standardized protocols. Include negative controls (no target RNA) in each run to establish baseline performance.
Response Measurement: For each replicate, record whether a positive response is detected based on a predetermined threshold (e.g., fluorescence intensity significantly above background). The result is binary (detected/not detected).
Data Analysis: Use logistic regression to model the probability of detection as a function of the target concentration:
- Let (xi) denote (\log2(ci)) where (ci) is the concentration
- Let (zi) be the number of detected values at concentration (ci)
- The logistic regression model assumes (zi) is binomially distributed with (fi = \frac{1}{1 + e^{-(\beta0 + \beta1 x_i)}}) [68]
LOD Calculation: Determine the concentration corresponding to a 95% detection probability from the fitted logistic regression curve. This represents the LOD [68].
Determining Limit of Quantification (LOQ)
The LOQ is the lowest concentration that can be quantified with acceptable precision and accuracy:
Precision Assessment: Calculate the coefficient of variation (CV) for replicates at each concentration level using the formula: (\exp(SD_{\ln\text{conc}}^2) - 1), assuming log-normal distribution of replicate concentrations [68].
Accuracy Assessment: Determine the percentage recovery for each concentration by comparing measured values to the known standard concentrations.
LOQ Establishment: Identify the lowest concentration where both CV ≤ 20-25% and recovery falls within 80-120% of the expected value [68]. This represents the LOQ for the toehold switch assay.
Characterizing Dynamic Range
The dynamic range for toehold switches spans from the LOQ to the maximum concentration where the dose-response relationship remains linear:
Dose-Response Curve: Test a wide range of target concentrations, from below the expected LOQ to concentrations where signal saturation occurs.
Linearity Assessment: Identify the concentration range where the dose-response relationship remains linear. The upper limit of quantification (ULOQ) is the highest concentration where the response remains linear without signal saturation.
ON/OFF Ratio Calculation: Calculate the dynamic range as the ON/OFF ratio—the fold-change between the maximum output signal (at saturation) and the basal signal (in the absence of trigger RNA) [1] [15]. High-performance toehold switches typically demonstrate ON/OFF ratios exceeding 400 [1].
Table 2: Experimental Replication Scheme for LOD Determination
| Concentration Level | Number of Replicates | Purpose |
|---|---|---|
| Blank (No template) | 20-30 | Establish baseline/false positive rate |
| Very Low (Near LOD) | 30-40 | Precisely characterize detection probability |
| Low (1-2x expected LOD) | 20-30 | Define lower end of logistic curve |
| Medium (3-5x expected LOD) | 15-20 | Establish middle range of logistic curve |
| High (>10x expected LOD) | 10-15 | Define upper asymptote of logistic curve |
Advanced Optimization Using Machine Learning
Traditional thermodynamic parameters have shown limited predictive value for toehold switch performance, with correlation metrics (R²) as low as 0.04-0.15 for predicting ON/OFF ratios [15]. Implementing machine learning approaches can significantly enhance design success:
Feature Extraction: Calculate comprehensive sequence and structure features including local base-pairing probabilities, minimum free energy, and codon usage bias that may impact co-transcriptional folding [12].
Model Training: Employ deep neural networks (DNNs) trained on large-scale toehold switch performance datasets. These models have demonstrated superior predictive capability (R² = 0.43-0.70) compared to traditional thermodynamic models [15].
Attention Visualization: Utilize techniques like VIS4Map (Visualizing Secondary Structure Saliency Maps) to identify important secondary structure patterns that correlate with successful switch function [15].
Iterative Design: Incorporate performance results from initial characterization into refined machine learning models to continuously improve LOD and dynamic range predictions for subsequent designs.
Machine Learning Optimization Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Toehold Switch Characterization
| Reagent/Category | Specific Examples | Function in Experimental Workflow |
|---|---|---|
| Toehold Switch Plasmids | pColADuet-1 vector [12] | Switch construction and maintenance |
| Target Expression Plasmids | pET15b vector [12] | Target RNA transcription |
| Cell-Free Expression System | PURExpress [15] | In vitro switch characterization |
| Fluorescent Reporters | GFP, mut3b-GFP with degradation tag [12] | Quantitative output measurement |
| RNA Production System | T7 RNA polymerase [12] | Target RNA synthesis |
| Computational Tools | NUPACK [13], ViennaRNA [13] | RNA secondary structure prediction |
| qPCR Reagents | ValidPrime assay, TaqMan probes [68] | Absolute quantification of targets |
| Fluorescent Dyes | EvaGreen, SYBR Green [69] [70] | Nucleic acid detection and quantification |
Establishing robust analytical performance parameters for toehold switches through rigorous determination of LOD and dynamic range is fundamental to their application in viral RNA detection. The protocols outlined herein provide a standardized framework for characterizing these essential parameters, incorporating both traditional statistical approaches and emerging machine learning methodologies. By implementing these comprehensive characterization workflows, researchers can advance toehold switch technology toward clinical application, enabling sensitive and reliable detection of viral pathogens across physiologically relevant concentration ranges. The integration of computational design with empirical validation represents the most promising path forward for developing next-generation RNA-based diagnostics with optimized performance characteristics.
Within the broader context of developing toehold switch technology for viral RNA detection, this document details standardized protocols for the clinical and field validation of these biosensors. A critical step in translating this synthetic biology tool from research to application is establishing its reliability against the current gold-standard method, reverse transcription quantitative polymerase chain reaction (RT-qPCR). This note provides a consolidated resource of validated experimental workflows and quantitative data, demonstrating how toehold switch-based diagnostics perform in direct correlation with RT-qPCR across human patient samples and agricultural plant specimens.
Toehold switch-based diagnostic systems have been validated for various targets, showing strong correlation with RT-qPCR results. The following table summarizes the key performance metrics from recent studies.
Table 1: Correlation of Toehold Switch-Based Diagnostics with RT-qPCR Results
| Target | Sample Type | Amplification Method | Detection Time | Limit of Detection (LoD) | Correlation with RT-qPCR | Citation |
|---|---|---|---|---|---|---|
| SARS-CoV-2 | Nasopharyngeal swab | NASBA | 60-100 min | 100 copies/sample | Efficient detection correlating with Ct values | [71] |
| SARS-CoV-2 | Nasopharyngeal swab | NASBA | 60-120 min | 1800 copies/sample | Successfully detected viral RNA from patient samples | [71] |
| SARS-CoV-2 | Saliva | RT-LAMP | 70 min | 120 copies/sample | N/A | [71] |
| Turnip Mosaic Virus (TuMV) | Pseudostellaria heterophylla plant crude RNA extracts | NASBA | 40 min - 90 min | 1 pM (40 min); <10 fM (90 min) | Detection confirmed in field samples; qRT-PCR confirmed high viral copies in infected leaves | [11] |
| West Nile Virus (WNV) | In vitro validation | N/A | N/A | N/A | Optimized RT-qPCR used for confirmation and quantification | [72] |
The provided data indicates that toehold switch sensors, particularly when coupled with an isothermal pre-amplification step like NASBA (Nucleic Acid Sequence-Based Amplification) or RT-LAMP (Reverse Transcription Loop-Mediated Isothermal Amplification), achieve a level of sensitivity that is applicable for real-world field and clinical diagnostics. The detection limits, reported in copies per sample or molar concentration, are functionally relevant for identifying active infections [71] [11].
Detailed Experimental Protocols
This section outlines step-by-step protocols for validating toehold switch performance against RT-qPCR in plant and human pathogen contexts.
Protocol for Plant Virus Detection (e.g., Turnip Mosaic Virus inPseudostellaria heterophylla)
This protocol is adapted from a 2025 study that successfully detected TuMV directly from both purified and crude RNA extracts of field samples [11].
Workflow Overview:
Materials & Reagents:
- Toehold Switch Plasmid: Contains switch sequence and downstream lacZ reporter gene.
- Cell-Free Protein Synthesis System: e.g., PURExpress or homemade extract.
- Trigger RNA / Target Amplicon: Product of NASBA amplification.
- Substrate: Chlorophenol red-β-D-galactopyranoside (CPRG).
- NASBA Primers: Designed for a conserved region of the TuMV genome (e.g., HC-pro or CP regions).
- qRT-PCR Kit: For independent validation.
Step-by-Step Procedure:
- Sample Collection and RNA Extraction:
- Collect leaf tissue from symptomatic and healthy plants in the field.
- Homogenize the tissue and perform RNA extraction using a commercial kit. For maximum field applicability, a crude extraction protocol (e.g., using simple buffers and centrifugation) can be validated alongside purified RNA extracts [11].
Isothermal Amplification (NASBA):
- Set up the NASBA reaction using the extracted RNA as template. NASBA is an isothermal RNA amplification technique that operates at 41°C, making it suitable for field use.
- Reaction Mix: Include primers, dNTPs, NTPs, and the enzymes AMV-RT, RNase H, and T7 RNA polymerase.
- Cycling: Incubate at 41°C for 90 minutes to amplify the target RNA sequence.
Toehold Switch Sensor Assay:
- Prepare the cell-free reaction mix containing the toehold switch plasmid, transcription-translation machinery, and the CPRG substrate.
- Add a defined volume of the NASBA amplicon (trigger RNA) to the cell-free reaction.
- Incubate at 37°C for a set period (e.g., 40-90 minutes) to allow for trigger-induced expression and enzymatic activity of the LacZ reporter protein.
Signal Detection and Validation:
- Visual Readout: Observe the color change from yellow to purple upon cleavage of CPRG by β-galactosidase. The time-to-color is proportional to the target concentration.
- Quantitative Readout: Use a microplate reader to measure the absorbance at 574 nm for more precise quantification.
- qRT-PCR Correlation: In parallel, analyze the original RNA samples using qRT-PCR to determine the cycle threshold (Ct) values. Plot the Ct values against the toehold switch sensor's response (time-to-color or end-point absorbance) to establish a correlation curve [11].
Protocol for Clinical SARS-CoV-2 Detection
This protocol synthesizes methods from multiple studies that developed toehold switches for COVID-19 diagnosis, correlating results with clinical RT-qPCR [71].
Workflow Overview:
Materials & Reagents:
- Toehold Switch System: Plasmid DNA containing the switch and a reporter gene (e.g., sfGFP for fluorescence or lacZ for colorimetry).
- Cell-Free Expression System: Lyophilized or liquid TX-TL system.
- Isothermal Amplification Kits: Commercial NASBA or RT-LAMP kits.
- RT-qPCR Kit: Approved for SARS-CoV-2 detection (targeting N, E, S, or RdRp genes).
Step-by-Step Procedure:
- Sample Processing:
- Obtain patient nasopharyngeal swab or saliva samples. Process swabs in viral transport media.
- Extract viral RNA using a commercial RNA extraction kit.
Target Amplification:
Toehold-Based Detection:
- Rehydrate a lyophilized cell-free reaction containing the toehold switch plasmid.
- Add a portion of the amplification product (NASBA or RT-LAMP amplicon) to the reaction.
- Incubate at 37°C for 1-2 hours.
- Readout: Measure fluorescence (for sfGFP) or colorimetric change (for lacZ). Results can be visualized by eye, camera, or microplate reader [71].
Correlation with Clinical RT-qPCR:
- Run the extracted RNA samples on a clinical RT-qPCR platform to determine the Ct value.
- A positive toehold switch result should correspond to samples with RT-qPCR Ct values below a specific threshold (e.g., Ct < 35), indicating a clinically relevant viral load. The PHANTOM system, for instance, showed efficient detection of viral RNA that correlated well with Ct values from RT-qPCR tests [71].
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key reagents and their functions essential for setting up toehold switch validation experiments.
Table 2: Essential Reagents for Toehold Switch Validation
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Toehold Switch Plasmid | The core biosensor element; encodes the riboregulator and reporter gene. | Second-generation design (tsgen2) minimizes sequence variability in structural regions [12]. |
| Cell-Free Protein Synthesis (CFPS) System | Provides the machinery for in vitro transcription and translation of the reporter. | PURExpress; TX-TL kits; lyophilized systems enable paper-based or field-stable formats [71]. |
| Isothermal Amplification Kit | Pre-amplifies target RNA to enhance detection sensitivity. | NASBA (for RNA) and RT-LAMP kits are widely used [71] [11]. |
| Reporter Substrates | Enable visual or fluorescent readout of sensor activation. | CPRG (for LacZ, colorimetric); ONPG (for LacZ, colorimetric) [71] [11]. |
| High-Fidelity Polymerase | Used for PCR during plasmid construction and trigger sequence verification. | Q5 Polymerase [12] [72]. |
Discussion
The protocols and data presented herein demonstrate that toehold switch biosensors, especially when integrated with isothermal amplification, provide a robust and rapid alternative to RT-qPCR for viral RNA detection in both clinical and agricultural settings. The strong correlation of results establishes the validity of this technology for use cases where speed, cost, and portability are critical.
Key to successful validation is the careful design of the toehold switch itself. Machine learning approaches, such as the VISTA framework, are now being employed to integrate features of both the sensor and the target RNA's structure, moving beyond traditional thermodynamic models to create more reliable designs [12] [15]. Furthermore, the use of versatile reporters like sfGFP and lacZ, coupled with smartphone-based colorimetry, paves the way for truly decentralized quantitative diagnostics [73].
Future work will focus on expanding these validation protocols to multiplexed detection platforms and streamlining the entire workflow into single-step, equipment-free devices, further bridging the gap between laboratory research and field-deployable diagnostic solutions.
Toehold switch technology represents a paradigm shift in synthetic biology, enabling the development of highly specific, programmable, and inexpensive RNA-sensing devices. These riboregulators are engineered to detect pathogen-specific RNA sequences, triggering a measurable signal upon activation. Their significance is profoundly evident in viral RNA detection, where they offer a promising alternative to costly standard methods like RT-qPCR. A critical driver for their adoption, especially in resource-limited settings or for large-scale screening programs, is their remarkably low production cost and the realistic potential for developing diagnostic tests priced under one dollar per unit. This application note details the experimental protocols and presents a structured cost-benefit analysis to guide researchers in leveraging this powerful technology.
Quantitative Cost-Benefit Analysis
The economic advantage of toehold switch-based diagnostics over conventional methods like RT-qPCR is substantial. The cost-saving arises from the minimal reagent requirements and the elimination of sophisticated, expensive equipment for signal amplification and detection.
Table 1: Cost and Performance Comparison of Viral RNA Detection Methods
| Method | Estimated Cost Per Reaction | Detection Time | Key Equipment | Limit of Detection |
|---|---|---|---|---|
| Toehold Switch (cell-free, paper-based) | $0.04 - $0.50 USD [71] | ~70 minutes - 2 hours [71] | Plate reader, incubator | ~120-1800 RNA copies [71] |
| Toehold Switch (with NASBA) | < $1.00 USD [71] | 60-100 minutes [71] | Microplate reader, incubator | 100 RNA copies [71] |
| RT-qPCR (Gold Standard) | ~$5.25 CAD (for extraction and test) [74] | Several hours (including sample processing) [71] | Real-time PCR cycler, RNA extraction equipment | Varies by protocol |
| Rapid Antigen Test (TR-Ag) | Lower than RT-PCR [75] | < 30 minutes [75] | None | Lower sensitivity than NAATs [75] |
The cost-benefit analysis extends beyond the direct reagent costs. Benefits include point-of-care applicability, long-term stability at room temperature when lyophilized on paper, and a rapid design-to-production cycle for emerging pathogens [71]. The primary costs involve initial sensor design, synthesis, and validation. For high-volume testing, the minimal variable cost makes toehold switches exceptionally cost-effective.
Experimental Protocols for Toehold Switch-Based Detection
The following protocol outlines the development of a toehold switch-based sensor for viral RNA, from design to signal detection.
Toehold Switch and Trigger RNA Design
Objective: To design a specific toehold switch riboregulator and its cognate trigger RNA sequence from a target viral genome (e.g., SARS-CoV-2).
Procedure:
- Target Selection: Identify a unique, conserved ~30-36 nt region within the viral RNA genome (e.g., from the S, N, or ORF1ab genes of SARS-CoV-2) to serve as the trigger sequence [71].
- Switch Design:
- Design the toehold switch RNA to include a hairpin structure that sequesters the Ribosome Binding Site (RBS) and start codon (AUG).
- The single-stranded toehold sequence at the 5' end must be perfectly complementary to the 5' end of the trigger RNA.
- The stem of the hairpin is designed to be complementary to the remaining portion of the trigger RNA. Early designs (1st generation) used variable complementarity across the entire stem to maximize diversity, while later designs (2nd generation) minimize variability in the upper stem [12] [76].
- Computational tools like NUPACK should be used to model RNA secondary structure and minimize off-target folding [12].
- Machine Learning Optimization: For enhanced performance, employ the Toehold-VISTA framework. This machine learning approach (Partial Least Squares Discriminant Analysis, PLS-DA) integrates sequence-structure features of both the sensor and the target RNA to predict and optimize sensor performance [12].
Plasmid Construction and Sensor Assembly
Objective: To clone the designed toehold switch and reporter gene into a plasmid for in vitro transcription-translation.
Procedure:
- Plasmid Backbone: Use a standard plasmid backbone (e.g., pCOLADuet-1) with a T7 promoter for in vitro expression [12] [77].
- Gene Synthesis: Synthesize the DNA fragment containing the toehold switch sequence followed by a reporter gene. Common reporter genes include:
- Cloning: Assemble the DNA fragment into the linearized plasmid using isothermal assembly methods (e.g., Gibson Assembly) [12].
- Transformation: Transform the assembled plasmid into chemically competent E. coli DH5α cells, plate on LB agar with appropriate antibiotics, and incubate overnight at 37°C [12] [77].
- Plasmid Purification: Pick single colonies, culture in liquid LB medium, and purify the plasmid using a commercial miniprep kit [12].
Cell-Free Expression and Detection
Objective: To produce the sensor output in a cell-free system and detect the presence of the target viral RNA.
Procedure:
- Cell-Free Reaction Setup: Reconstitute a commercial cell-free transcription-translation (IVTT) system according to the manufacturer's instructions.
- Sensor Application: Immobilize the purified plasmid DNA onto a paper disc or add it directly to the liquid IVTT reaction mixture [71].
- Sample Addition: Add the extracted viral RNA (or an isothermally pre-amplified sample, see section 3.4) to the reaction.
- Incubation: Incubate the reaction at 37°C for 60-120 minutes to allow for trigger binding, switch activation, and reporter protein production [71].
- Signal Detection:
- For Fluorescent Reporters (sfGFP): Measure fluorescence intensity using a microplate reader (Ex/~485 nm, Em/~510 nm). A positive signal indicates viral RNA detection [71].
- For Colorimetric Reporters (lacZ): Add the substrate CPRG. The development of a red color indicates a positive detection, which can be visualized by the naked eye, a camera, or quantified with a plate reader [71] [16].
Optional: Pre-amplification of Viral RNA
Objective: To increase detection sensitivity by amplifying the target RNA sequence prior to detection.
Procedure:
- RNA Extraction: Extract viral RNA from patient samples (e.g., nasopharyngeal swab or saliva) using a cost-effective method, such as a modified TRIzol-based protocol, which can reduce cost to ~$0.58 per sample [74].
- Isothermal Amplification: Perform an isothermal amplification step to increase the number of trigger RNA molecules.
- Detection: Use the amplified product as the input for the cell-free detection system described in section 3.3.
Workflow Visualization
The following diagram illustrates the logical and experimental workflow for toehold switch-based viral RNA detection.
Diagram 1: Toehold Switch Viral Detection Workflow
The molecular mechanism of the toehold switch is a key determinant of its function and cost-effectiveness.
Diagram 2: Toehold Switch Activation Mechanism
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Materials for Toehold Switch Experiments
| Item | Function/Description | Example/Note |
|---|---|---|
| Toehold Switch Plasmid | DNA template encoding the riboregulator and reporter gene. | Designed in-silico; cloned into a vector with a T7 promoter [12]. |
| Cell-Free Protein Synthesis System | In vitro transcription-translation mixture. | Commercial E. coli lysate systems (e.g., PURExpress) are commonly used [71]. |
| Reporter Gene Construct | Encodes the detectable output protein. | sfGFP (fluorescence) or lacZ (colorimetric) are widely used [71] [16]. |
| Colorimetric Substrate | Enzyme substrate for visual readout. | Chlorophenol red-β-D-galactopyranoside (CPRG) for lacZ, turns from yellow to red [71]. |
| Isothermal Amplification Kits | For target RNA pre-amplification to enhance sensitivity. | Kits for NASBA or RT-LAMP [71]. |
| Solid Support | Platform for portable, stable sensor deployment. | Whatman filter paper or other porous materials for lyophilizing the reaction [71]. |
The rapid and accurate detection of viral pathogens remains a cornerstone of effective public health responses. Within the realm of molecular diagnostics, technologies such as Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) have long been the gold standard. However, the field is rapidly evolving with the emergence of novel, programmable nucleic acid detection tools. Among these, toehold switch sensors and CRISPR-Cas-based systems represent a new generation of diagnostics that offer potential for rapid, sensitive, and equipment-free testing. This Application Note provides a comparative analysis of these three key technologies—toehold switches, CRISPR-Cas, and RT-qPCR—framed within the context of advancing research into toehold switches for viral RNA detection. We summarize their core characteristics, present detailed experimental protocols, and visualize their workflows to aid researchers and drug development professionals in selecting and implementing the most appropriate method for their specific applications.
Technology Comparison at a Glance
The following table provides a high-level comparison of the core characteristics, advantages, and limitations of toehold switch sensors, CRISPR-Cas systems, and RT-qPCR.
Table 1: Core Characteristics of Toehold Switches, CRISPR-Cas, and RT-qPCR
| Feature | Toehold Switch Sensors | CRISPR-Cas Diagnostics | RT-qPCR |
|---|---|---|---|
| Core Principle | RNA-based riboregulator; target binding induces conformational change to permit translation of a reporter gene [4] [14]. | crRNA-guided Cas protein (e.g., Cas12, Cas13) binds target, activating collateral cleavage of a reporter molecule [78] [79]. | Enzyme-driven amplification of cDNA with real-time fluorescent quantification. |
| Primary Target | RNA [4] [14]. | DNA (Cas12) or RNA (Cas13) [78]. | RNA (via initial reverse transcription). |
| Typical Readout | Colorimetric (e.g., β-galactosidase/LacZ) [14], fluorescent. | Fluorescent, visual (lateral flow) [78] [80]. | Fluorescent (real-time). |
| Sensitivity | ~120 copies of RNA (with RT-LAMP amplification) [14]. | Attomolar (aM) levels; as low as 0.11 copies/μL with advanced methods [79] [80]. | High; typically down to a few copies/μL [81]. |
| Key Advantage | Fully synthetic, programmable; can be freeze-dried for paper-based tests [4] [14]. | Exceptional sensitivity and specificity; versatile readouts; rapid (results in ~30-40 min) [78] [79] [80]. | Gold standard; high throughput; quantitative; well-established protocols and regulations. |
| Key Limitation | Often requires separate amplification step for high sensitivity [14]. | Can require optimized pre-amplification; potential for off-target effects [78] [80]. | Requires sophisticated thermocyclers and labs; skilled personnel; longer time-to-result [81] [82]. |
Detailed Experimental Protocols
Protocol: Toehold Switch Sensor for Coronavirus Detection
This protocol, adapted from Kim et al. (2021), details the use of a toehold switch sensor coupled with modified RT-LAMP for the colorimetric detection of coronaviruses like SARS-CoV-2 [14].
1. Design and In Vitro Validation of Toehold Switch:
- Toehold Switch Design: Design the toehold switch and its cognate trigger RNA sequence using NUPACK software to optimize the normalized ensemble defect (NED), ensuring a low percentage of incorrectly paired nucleotides at equilibrium [14].
- Sensor Screening: Clone the selected toehold switch sequence upstream of a reporter gene (e.g., lacZ). Test the sensor in vitro by incubating with and without synthetic trigger RNA. Measure the absorbance at 570 nm after adding the substrate CPRG. Select sensors with high fold-change (on/off ratio) and low background signal [14].
2. Sample Preparation and Modified RT-LAMP Amplification:
- RNA Extraction: Extract viral RNA from patient samples (e.g., nasopharyngeal swabs) using a standard silica-column or magnetic-bead based method.
- Primer Design for RT-LAMP: Design LAMP primers (F3, B3, FIP, BIP) targeting a conserved region of the viral genome (e.g., SARS-CoV-2 N gene).
- T7 Promoter Incorporation: Incorporate a T7 RNA polymerase promoter sequence into the 5' end of the FIP primer [14].
- Amplification Reaction: Perform the RT-LAMP reaction in a single tube. The reaction mix typically contains:
- Target RNA template
- LAMP primer mix (including the T7-modified FIP primer)
- WarmStart RTx Reverse Transcriptase
- Bst 2.0/3.0 DNA Polymerase (strand-displacing)
- dNTPs
- Isothermal amplification buffer (e.g., with MgSO₄)
- Incubation: Incubate the reaction at 65°C for 30-60 minutes to generate amplicons. The T7 promoter incorporated into the amplicons allows for subsequent transcription [14].
3. Toehold Reaction and Colorimetric Detection:
- Transcription: Following LAMP amplification, add T7 RNA polymerase directly to the same tube to transcribe the DNA amplicons into RNA. These RNA transcripts serve as the triggers for the toehold switch.
- Cell-Free Protein Synthesis (CFPS): Combine the transcription reaction with a CFPS system (e.g., E. coli lysate) containing:
- The engineered toehold switch plasmid DNA
- Ribonucleotides (rNTPs)
- amino acids
- Energy regeneration system (e.g., phosphoenolpyruvate)
- Colorimetric Reporting: Include the substrate Chlorophenol red-β-D-galactopyranoside (CPRG) in the CFPS mix.
- Incubation and Visualization: Incubate the one-pot reaction at 37°C for 60-90 minutes. A positive detection event unfolds the toehold switch, allowing translation of β-galactosidase (LacZ), which hydrolyzes yellow CPRG to purple Chlorophenol red (CPR). The color change can be monitored visually or quantified by measuring absorbance at 570 nm [14].
Protocol: One-Pot RPA-CRISPR/Cas12a Assay
This protocol, based on work by Huang et al. (2024) and others, describes a one-pot assay for specific DNA target detection, which can be adapted for viral cDNA by including an initial reverse transcription step [83] [79].
1. crRNA Design and Optimization:
- crRNA Design: Design crRNAs to target a specific, conserved region of the pathogen's genome. The crRNA should be ~20 nucleotides complementary to the target adjacent to a PAM sequence (TTTV for Cas12a).
- Optimization: Systematically optimize the concentration of crRNA (e.g., ~133 nM) and the Cas12a-to-crRNA ratio (e.g., 1:1) for maximal activity and minimal background [83].
2. One-Pot Reaction Setup:
- Reagent Preparation: Prepare two separate mixes in a single tube, physically separated (e.g., the RPA mix in the bottom and the CRISPR detection mix on the lid).
- RPA Mix (Bottom): This contains:
- Template DNA (or RNA/cDNA for viral detection)
- RPA forward and reverse primers
- Rehydration buffer
- Magnesium acetate (to initiate amplification)
- CRISPR Detection Mix (Lid): This contains:
- Cas12a enzyme
- Optimized crRNA
- Fluorescent single-stranded DNA (ssDNA) reporter (e.g., FAM-TTATT-BHQ1)
- RPA Mix (Bottom): This contains:
- Amplification and Detection: Centrifuge the tube briefly to combine the mixes. Incubate at 37°C for 20 minutes for RPA amplification. The amplicons then activate the Cas12a/crRNA complex, which collaterally cleaves the ssDNA reporter, generating a fluorescent signal [83].
3. Result Interpretation:
- Fluorescence Detection: After a total reaction time of ~30 minutes, visualize the tube under blue or UV light. Bright fluorescence indicates a positive result.
- Quantification (Optional): For quantitative results, measure fluorescence intensity in real-time using a plate reader or a portable fluorometer [83].
Workflow Visualization
The following diagrams illustrate the core signaling pathways and experimental workflows for the two primary protocols described above.
Toehold Switch with RT-LAMP Workflow
Diagram Title: Toehold Switch Detection Path
RPA-CRISPR/Cas12a Detection Mechanism
Diagram Title: CRISPR-Cas12a Detection Path
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of these diagnostic platforms relies on a core set of reagents and materials. The following table lists essential items for setting up toehold switch and CRISPR-Cas assays.
Table 2: Essential Research Reagents for Novel Diagnostic Assays
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing DNA polymerase for isothermal amplification (LAMP). | Amplifying target viral RNA sequences in the RT-LAMP step of the toehold switch protocol [14]. |
| T7 RNA Polymerase | Bacteriophage-derived RNA polymerase that transcribes DNA templates into RNA. | Generating RNA trigger strands from RT-LAMP amplicons in the toehold switch assay [14]. |
| Cas12a (Cpf1) Enzyme | CRISPR-associated protein that, upon crRNA-guided target DNA recognition, exhibits non-specific single-stranded DNA (ssDNA) cleavage (collateral activity). | The core effector protein in the RPA-CRISPR/Cas12a assay for specific target detection and signal generation [83]. |
| crRNA | CRISPR RNA; a short synthetic RNA that guides the Cas protein to a specific DNA target sequence. | Directing Cas12a to the pathogen-specific amplicon in the CRISPR detection assay [83]. |
| Recombinase Polymerase | Enzyme complex that facilitates strand invasion and primer binding for isothermal amplification in RPA. | Rapidly amplifying target DNA at a constant temperature (37-42°C) in the RPA-CRISPR/Cas12a assay [83]. |
| Fluorescent ssDNA Reporter | A short ssDNA oligonucleotide labeled with a fluorophore and a quencher; cleavage separates the pair, generating a signal. | Detecting the collateral cleavage activity of activated Cas12a in real-time or endpoint fluorescence readouts [79] [83]. |
| Cell-Free Protein Synthesis System | A lysate-based system (e.g., from E. coli) containing the machinery for transcription and translation outside a living cell. | Expressing the reporter protein (e.g., LacZ) from the unfolded toehold switch in a test tube [14]. |
| Toehold Switch Plasmid | A DNA vector encoding the engineered RNA toehold switch sequence upstream of a reporter gene. | The core sensing element in the toehold switch assay; its sequence dictates target specificity [14]. |
Toehold switch sensors represent a significant advancement in synthetic biology, enabling the programmable detection of specific RNA sequences. Within viral diagnostics, their operational advantages—namely rapid detection, minimal equipment requirements, and true field-deployability—are transforming approaches to outbreak management and point-of-care testing [11]. These riboregulators function through a conformational change upon binding to a target viral RNA trigger, which exposes a ribosome binding site (RBS) and initiates translation of a reporter gene [16]. This direct mechanism bypasses complex instrumentation, forming the basis for highly portable and rapid diagnostic platforms. This application note details quantitative performance data and provides standardized protocols to leverage these operational advantages in viral RNA detection research.
Quantitative Operational Advantages of Toehold Switches
The utility of toehold switches in viral detection is quantified by their speed, sensitivity, and minimal hardware dependencies. The following tables summarize key performance metrics from recent applications.
Table 1: Performance Metrics for Toehold Switch-Based Viral Detection Platforms
| Target Virus / Application | Detection Limit | Time to Result | Key Equipment | Reference |
|---|---|---|---|---|
| Turnip Mosaic Virus (TuMV) | 10 fM (after 90 min) | ~40 min (for 1 pM) to 90 min | Cell-free system, NASBA amplification [11] | |
| Zika & Novel Coronaviruses | Not Specified | Rapid | Paper-based strip, cell-free system [16] | |
| General Toehold Switch Function | N/A | N/A | Core Components: In vitro transcription-translation system, colorimetric/fluorimetric reporter [11] [16] |
Table 2: Comparison of Detection Modalities for Toehold Switches
| Readout Method | Reported Molecule | Equipment Needs | Best Suited For |
|---|---|---|---|
| Colorimetric | LacZ (β-galactosidase) | Visual assessment, optional plate reader [11] [16] | Low-resource, field-deployable assays |
| Fluorimetric | GFP (e.g., eGFP, mut3b-GFP) | Flow cytometer, fluorimeter [12] [10] | Laboratory-based quantification and high-throughput screening |
Experimental Protocols for Field-Deployable Viral Detection
Protocol: Paper-Based Toehold Switch Sensor for Viral RNA
This protocol outlines the use of a paper-based cell-free system coupled with a colorimetric readout, ideal for field deployment [16].
Principle: Viral RNA triggers the toehold switch, leading to the synthesis of β-galactosidase. This enzyme cleaves a yellow substrate (CPRG) into a red product, providing a visual readout [16].
Key Reagents and Materials:
- Toehold Switch Plasmid DNA: Encoding the switch and the LacZ reporter gene.
- Cell-Free Protein Synthesis System: A commercially available or homemade E. coli-based transcription-translation mix.
- Whatman Grade 1 Filter Paper.
- Substrate Solution: 2.5 mM Chlorophenol-red-β-D-galactopyranoside (CPRG) in DMSO.
- RNA Sample: Purified or crudely extracted viral RNA.
Procedure:
- Spotting and Immobilization: Apply the cell-free reaction mix, pre-mixed with the toehold switch plasmid DNA, onto the filter paper. Air-dry the spotted paper completely.
- Reaction Initiation: Apply the RNA sample mixed with the CPRG substrate solution directly onto the pre-spotted and dried paper.
- Incubation and Detection: Incubate the paper at 37°C for 30-90 minutes. Observe for a color change from yellow to red, indicating the presence of the target viral RNA.
Protocol: Toehold Switch Sensor Coupled with NASBA Amplification
This protocol describes a highly sensitive method that combines Nucleic Acid Sequence-Based Amplification (NASBA) with a toehold switch sensor for detecting low-abundance viral targets, such as TuMV, in plant samples [11].
Principle: The viral RNA target is first amplified isothermally via NASBA. The resulting amplicons then act as triggers for the toehold switch in a cell-free system, with detection via a colorimetric LacZ readout [11].
Key Reagents and Materials:
- NASBA Primers: Designed to target a conserved region of the viral genome (e.g., the HC-pro region for TuMV).
- NASBA Enzyme Mix: Contains T7 RNA polymerase, RNase H, and avian myeloblastosis virus (AMV) reverse transcriptase.
- Toehold Switch Sensor: Designed in silico using tools like MeFit Toehold Designer and the ViennaRNA package, targeting the NASBA amplicon [11].
- Cell-Free Expression System.
Procedure:
- RNA Extraction: Extract total RNA from the sample (e.g., plant leaf tissue) using a commercial kit or a crude extraction buffer suitable for field use.
- NASBA Amplification:
- Prepare the NASBA reaction mix containing primers, dNTPs, NTPs, and the enzyme mix.
- Add the extracted RNA template.
- Incubate at 41°C for 90 minutes.
- Toehold Switch Detection:
- Combine the NASBA amplicon with the cell-free reaction mix containing the toehold switch plasmid.
- Add the CPRG substrate.
- Incubate at 37°C and monitor the color change. The entire process, from RNA extraction to result, can be completed in approximately 3 hours [11].
Visualizing Toehold Switch Workflows and Signaling
Toehold Switch Mechanism and Workflow
Toehold Switch Molecular Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Toehold Switch Research
| Reagent/Material | Function/Description | Example Use Case |
|---|---|---|
| Toehold Switch Plasmid | Engineered vector containing the riboregulator and reporter gene (e.g., LacZ, eGFP). | Core sensing element in cell-free systems or transfected into cells [10] [11]. |
| Cell-Free Protein Synthesis System | Lysate-based system (e.g., from E. coli) capable of transcription and translation in vitro. | Enables reaction setup without live cells, crucial for field-deployable diagnostics [11] [16]. |
| Isothermal Amplification Mix (NASBA/RPA) | Enzyme mix for amplifying RNA/DNA targets at a constant temperature. | Boosts sensitivity for low-abundance viral targets; avoids the need for thermal cyclers [11]. |
| Colorimetric Substrate (CPRG) | Chromogenic enzyme substrate (yellow to red upon cleavage by β-galactosidase). | Provides a visual, equipment-free readout for presence of target [11] [16]. |
| Fluorescent Reporter Plasmid | Plasmid encoding a fluorescent protein (e.g., eGFP, mCherry) under toehold switch control. | Allows for quantitative measurement via flow cytometry or fluorimetry in lab settings [12] [10]. |
Conclusion
Toehold switch technology represents a paradigm shift in molecular diagnostics, offering a highly programmable, sensitive, and inexpensive platform for viral RNA detection. By integrating foundational RNA design principles with robust methodological applications and sophisticated signal amplification strategies, this platform meets critical needs for rapid and deployable testing. Future directions are poised to expand its utility through multiplexed detection of several pathogens in a single assay, integration into wearable sensors, and adaptation into therapeutic delivery systems. For researchers and drug developers, mastering this technology opens avenues for creating next-generation diagnostics that are not only crucial for pandemic preparedness but also for routine monitoring of infectious diseases in both clinical and resource-limited settings.
References
- 1. Toehold Switches: De-Novo-Designed Regulators of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4265554/]
- 2. Riboswitch-inspired toehold for gene regulation in... riboregulators [https://pmc.ncbi.nlm.nih.gov/articles/PMC9071393/]
- 3. Toehold Switch Plus Signal Amplification Enables Rapid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10840733/]
- 4. RNA-Based Sensor Systems for Affordable Diagnostics in ... [https://www.sciencedirect.com/org/science/article/pii/S2161506324001554]
- 5. Toehold switch assembly [https://www.protocols.io/view/toehold-switch-assembly-72bhqan]
- 6. Sequence-to-function deep learning... | Nature Communications [https://www.nature.com/articles/s41467-020-18676-2?error=cookies_not_supported]
- 7. Intelligent guide RNA: dual toehold switches for modulating ... [https://www.nature.com/articles/s42003-024-06988-8]
- 8. Developments of Riboswitches and Toehold Switches ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7247568/]
- 9. Single-molecule force spectroscopy of toehold-mediated ... [https://www.nature.com/articles/s41467-024-51813-9]
- 10. Results | Warsaw - iGEM 2025 [https://2025.igem.wiki/warsaw/results]
- 11. Detection of TuMV by a toehold switch sensor coupled with ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-025-01394-5]
- 12. Toehold-VISTA: A machine learning approach to decipher ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363951/]
- 13. Engineering Toehold-Mediated Switches for Native RNA ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283622002819]
- 14. Detection of Coronaviruses Using RNA Toehold Switch ... [https://www.mdpi.com/1422-0067/22/4/1772]
- 15. A deep learning approach to programmable RNA switches [https://www.nature.com/articles/s41467-020-18677-1]
- 16. A paper-based RNA sensor turns red to reveal hidden ... [https://www.nature.com/articles/d44151-025-00130-1]
- 17. NUPACK 4 User Guide [https://docs.nupack.org/]
- 18. Model Specification [https://docs.nupack.org/model/]
- 19. Comparative analysis of RNA secondary structure accuracy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10473517/]
- 20. Toehold designer | WageningenUR - iGEM 2024 [https://2024.igem.wiki/wageningenur/toehold-model]
- 21. RNA Toehold Switch Optimization - NeuroSplice [https://2025.igem.wiki/calucsf/results]
- 22. Evolution shapes and conserves genomic signatures in ... [https://www.nature.com/articles/s42003-024-07098-1]
- 23. A novel approach to finding conserved features in low ... [https://www.nature.com/articles/s41598-023-39207-1]
- 24. Comparative Bioinformatic Analysis Reveals Conserved ... [https://www.mdpi.com/1422-0067/25/11/5764]
- 25. Global landscape of SARS-CoV-2 mutations and conserved ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-023-03996-w]
- 26. Engineering CRISPR guide RNAs for programmable RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10754282/]
- 27. The Role of RNA Secondary Structure in Regulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8346122/]
- 28. Engineered RNA biosensors enable ultrasensitive SARS- ... [https://www.life-science-alliance.org/content/4/12/e202101213]
- 29. Autocatalytic base editing for RNA-responsive translational ... [https://www.nature.com/articles/s41467-023-36851-z]
- 30. A comparison of experimental assays and analytical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9288987/]
- 31. Compact RNA sensors for increasingly complex functions ... [https://www.nature.com/articles/s41557-025-01907-8]
- 32. Isothermal Amplification [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/isothermal-amplification?srsltid=AfmBOopHH86vcyYZ-op8nDa81fI9XUvmtn7bxAIiX2May0pbH3ykEL8S]
- 33. A rapid and high sensitivity RNA detection based on ... [https://www.nature.com/articles/s41598-022-14107-y]
- 34. Rapid and accurate clinical testing for COVID-19 by nicking ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566321007260]
- 35. Real-Time Reverse Transcription Loop-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC321710/]
- 36. CRISPR/Cas13a analysis based on NASBA amplification ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914024011044]
- 37. Isothermal Amplification Techniques Gain Momentum [https://www.meridianbioscience.com/lifescience-blog/isothermal-amplification-techniques-gain-momentum/]
- 38. An improved nucleic acid sequence-based amplification ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0265391]
- 39. Detection of Coronaviruses Using RNA Toehold Switch ... [https://pubmed.ncbi.nlm.nih.gov/33578973/]
- 40. Comparison between RT-PCR, NASBA and RT-LAMP Methods for Detection of Coxsackievirus B3 [https://www.bmmj.org/article_32030.html]
- 41. A sensitive mNeonGreen reporter system to measure ... [https://www.nature.com/articles/s42003-020-01375-5]
- 42. In vivo selection of sfGFP variants with improved and reliable ... [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-017-1008-5]
- 43. Green cGMP Protocol [https://montanamolecular.com/protocols/mneongreen-cgmp-protocol/?srsltid=AfmBOopCIYx82PA0YLYV_z9yQGOrTaXbhXHzz2FwSo1re1mvvIDBJiw1]
- 44. A paper-based, cell-free biosensor system for the detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6402643/]
- 45. Toehold Switches for Synthetic Biology - Wyss Institute [https://wyss.harvard.edu/technology/toehold-switches-for-synthetic-biology/]
- 46. An Update on RNA Virus Discovery: Current Challenges and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300815/]
- 47. Simultaneous detection of respiratory virus RNA on ... [https://www.nature.com/articles/s41598-025-22296-5]
- 48. Breakthrough in RNA-targeted drug discovery offers hope ... [https://www.news-medical.net/news/20251006/Breakthrough-in-RNA-targeted-drug-discovery-offers-hope-against-viral-diseases.aspx]
- 49. Toehold switch based biosensors for sensing the highly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8976095/]
- 50. Towards detection of SARS-CoV-2 RNA in human saliva: A ... [https://www.sciencedirect.com/science/article/pii/S1871678421000893]
- 51. PLANT-Dx: A Molecular Diagnostic for Point-of-Use ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6479721/]
- 52. Directed evolution improves the catalytic efficiency of TEV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7004888/]
- 53. Engineering the Tobacco Etch Virus Protease toward a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12455634/]
- 54. Recent advances in cascade isothermal amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10156408/]
- 55. Toehold Switch | DUT-China - iGEM 2025 [https://2025.igem.wiki/dut-china/toehold_switch]
- 56. Kinetics and Activation Strategies in Toehold-Mediated and ... [https://www.mdpi.com/2079-6374/15/10/683]
- 57. Decentralizing Cell-Free RNA Sensing With the Use of Low ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.727584/full]
- 58. Detection of TuMV by a toehold switch sensor coupled with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12147267/]
- 59. Inexpensive and colorimetric RNA detection at ambient ... [https://biotechsustainablematerials.biomedcentral.com/articles/10.1186/s44316-024-00007-w]
- 60. RNA Quality Control Using External Standard RNA - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7256816/]
- 61. Lyophilization Development: Room Temp Biologics Stability [https://www.leukocare.com/blog/lyophilization-development-for-room-temperature-biologics]
- 62. Freeze-Drying of mRNA-LNPs Vaccines: A Review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389932/]
- 63. Lyophilization Formulation for Gene Therapy Stability [https://www.leukocare.com/blog/lyophilization-formulation-for-gene-therapy-products]
- 64. From pilot to production: Scaling lyophilization with ... [https://www.pharmaceutical-technology.com/sponsored/from-pilot-to-production-scaling-lyophilization-with-confidence/]
- 65. Practical advice in the development of a lyophilized protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11744310/]
- 66. Lyophilization process optimization and molecular ... [https://www.nature.com/articles/s41541-023-00732-9]
- 67. Enhancing Biopharmaceutical Stability Through ... [https://www.pharmasalmanac.com/articles/enhancing-biopharmaceutical-stability-through-lyophilization]
- 68. Methods to determine limit of detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5496743/]
- 69. What is the Difference Between Dye-Based and Probe-Based ... [https://shop.cgenomix.com/archives/7467/what-is-the-difference-between-dye-based-and-probe-based-systems-in-qpcr-enzymes/?srsltid=AfmBOoqtG_BRrxkbnU8YXKUARVap_hypi_BvO260yp6pU93v7bqNIwCm]
- 70. Should I use probe-based detection or dye ... [https://www.aatbio.com/resources/faq-frequently-asked-questions/should-i-use-probe-based-detection-or-dye-based-detection-for-my-qpcr-analysis]
- 71. Update on the Development of Toehold Switch-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9110250/]
- 72. Development of Toehold Switches as a Novel Ribodiagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9859090/]
- 73. Measurement | Baltimore-BioCrew - iGEM 2025 [https://2025.igem.wiki/baltimore-biocrew/measurement]
- 74. A cost-effective RNA extraction and RT-qPCR approach to ... [https://www.nature.com/articles/s41598-024-52534-1]
- 75. Cost-effectiveness analysis of COVID-19 tests in the unified ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10498608/]
- 76. Toehold-switch architecture selection [https://bio-protocol.org/exchange/minidetail?id=8511575&type=30]
- 77. Toehold Switch‐Based Approach for Engineering Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146417/]
- 78. CRISPR‐driven diagnostics: Molecular mechanisms, clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464570/]
- 79. Ultrasensitive detection of clinical pathogens through a ... [https://www.nature.com/articles/s41467-025-59219-x]
- 80. Recent developments and future directions in point-of-care ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11717804/]
- 81. A Critical Review of the CRISPR-Cas Technology in the ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12267969/]
- 82. Progress in the application of isothermal amplification ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1601644/full]
- 83. Comparative Evaluation of PCR-Based, LAMP and RPA- ... [https://www.mdpi.com/1422-0067/25/11/5773]